参考文献:黎海芪. 实用儿童保健学(第2版)[M]. 北京:人民卫生出版社,2022.
❝本文为关于儿童眼发育异常(包括形态畸形、先天性结构异常及相关综合征)的系统学习记录,仅为个人知识整理。
开篇总述
眼、耳、鼻的发育异常常与儿童情感、智力、体格发育异常及特殊面容相关。这是因为面部发育与前额、脊索前板发育密切相关。儿科与儿童保健医生在临床体格检查时,需仔细观察眼、耳、鼻的发育情况——异常的面部发育,往往是提示潜在综合征的重要线索。
一、眼形态异常或畸形
重点关注睑裂、眼距、眼睑、睫毛、虹膜、角膜、巩膜等眼外部结构。这类异常在儿童中较常见,常伴随特殊面容,是临床筛查的关键切入点。
(一)睑裂相关异常
- 睑裂倾斜度异常:表现为睑裂向外上方倾斜,是21-三体综合征的典型特殊面容之一。
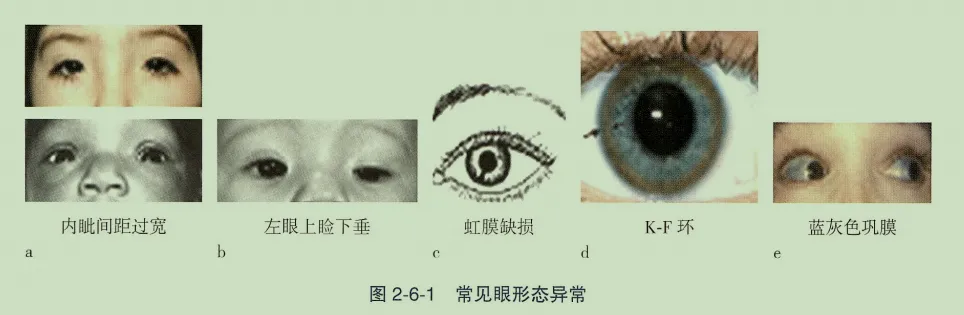
- 内眦间距过宽:指内眦间骨性间距增加,常伴内眦赘皮和小脸裂。需与“眼距过宽”区分,后者仅为眼裂间距宽,无内眦赘皮或小脸裂。可见于21-三体、Noonan综合征等多种伴特殊面容的综合征。
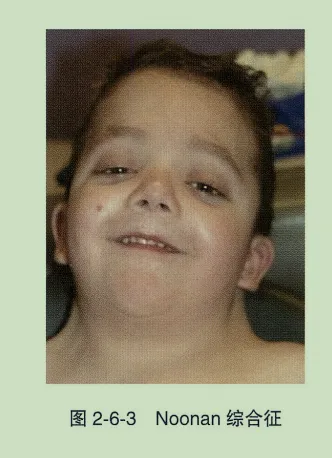
- 睑裂短小:睑裂横向或纵向径线小于正常,是胎儿酒精谱系障碍的典型表现之一,常同时伴小头畸形、鼻梁低、内眦赘皮、小耳畸形、下颌小等特征。
(二)眼睑异常
1. 上睑下垂
- 程度与表现:轻者仅部分遮盖瞳孔,重者可完全遮盖瞳孔。双侧下垂者视物时习惯仰首以克服视力障碍。
- 发病特点:多为双侧,也可单侧,呈常染色体显性或隐性遗传。
- 关联综合征:可见于Dubowitz综合征、Neu-Laxova综合征、Noonan综合征、Freeman-Sheldon综合征等。
2. 眼睑其他异常
- 倒睫:常伴随睑内翻出现,长期摩擦角膜可引发结膜充血、角膜损伤。
- 睑缘异常:如Zellweger综合征可出现眼外眦赘皮,同时伴眼部多部位受累。
(三)虹膜、角膜、巩膜异常
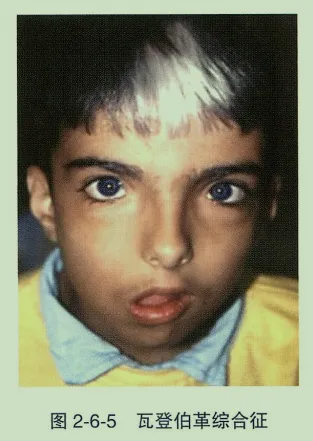
- 虹膜异色:表现为单侧或片状色素减退,由先天性虹膜基质发育不全所致。一般不影响视力,多为良性表现,常见于瓦登伯革综合征。
- 虹膜缺损:虹膜下方缺损,严重者可表现为无虹膜。由胚胎发育过程中胚裂闭合不全所致。可孤立出现,或并发于Wilms瘤综合征及其他泌尿生殖系统综合征。
- K-F环(角膜色素环):角膜后弹力层出现铜沉积形成的色素环,是肝豆状核变性的重要体征。多出现神经症状时需排查,需用裂隙灯检查确诊。
- 蓝灰色巩膜:巩膜呈蓝灰色改变,见于成骨不全症,由胶原合成异常致骨组织脆弱所致,常伴易骨折、骨骼畸形等全身表现。
(四)睫毛异常
主要表现为倒睫,即睫毛倒向眼球方向,常伴随睑内翻出现。需与单纯睑内翻鉴别:倒睫多为睫毛位置异常,而睑内翻是眼睑边缘向内翻转。
二、先天性眼发育异常
胚胎早期(受孕至胎儿娩出前)受感染、有害射线、毒物等伤害,或遗传因素影响,可导致眼组织发育受阻,引发各类先天性眼病。多在出生时即存在,部分随生长发育逐渐显现。
(一)晶状体异常
- 晶状体混浊:包括白内障、晶状体部分混浊、晶状体密度异常。可遮挡光线进入眼内,致视力下降,婴幼儿可引发形觉剥夺性弱视。
- 晶状体大小与形态异常:包括小晶状体、球形晶状体、晶状体异位等。易伴随屈光不正和斜视,严重者可致眼压升高。
- 晶状体发育不全:晶状体结构发育不完善,伴功能异常,多与额鼻发育不良综合征等遗传综合征相关。
(二)胚裂闭合不全
由胚眼发育过程中胚裂闭合障碍所致,可累及虹膜、睫状体、脉络膜、视网膜,引发对应组织缺损。常与先天性小眼球、视网膜发育不良并发,部分合并全身多系统畸形。
(三)瞳孔残膜
又称永存瞳孔膜。胎儿7-9月龄时,正常晶状体前囊膜的瞳孔残膜应萎缩吸收,若未完全吸收则遗留。轻者无明显症状,重者可遮挡瞳孔区影响视力发育。需与瞳孔粘连鉴别,后者多为炎症后虹膜与晶状体粘连,无残膜组织。
(四)角膜异常
- 常见类型:先天性大角膜(角膜横径>13mm)、先天性小角膜(角膜横径<10mm,常伴眼球较小)、扁平角膜、球形角膜、巩膜化角膜、先天性角膜混浊、角膜皮样瘤等。
- 临床意义:角膜混浊可直接致视力障碍;角膜皮样瘤多为先天性良性肿瘤,随年龄增长可能增大,需尽早评估手术时机。
- 关联综合征:额鼻发育不良综合征、Alport综合征、Apert综合征等均可能伴随角膜异常。
(五)先天性小眼球
指眼球体积显著小于正常,可合并眼球缺如。常伴眼前节发育不全、先天性白内障、脉络膜视网膜缺损、视网膜发育不良。与PAX6、SOX10、MITF、MCOP、NNO2等基因突变相关。临床需评估视力发育和眼球结构,必要时安装义眼座改善外观,同时排查全身合并畸形。
(六)视神经发育不良
表现为视神经纤维层发育不全,致视力下降、视野缺损。约13%受累患者存在垂体功能异常,约1/4患者可见透明隔部分或全部消失。视神经发育不良合并透明隔发育不全,称为DeMosier综合征。临床需定期监测视力、视野,同时排查内分泌异常(如生长激素缺乏、甲状腺功能异常)。
三、与眼发育异常相关的综合征与基因
眼发育异常多为全身综合征的眼部表现,单个或多个基因缺陷、染色体结构畸变或数目异常,均可能导致眼发育畸形,需结合基因检测和全身表现明确诊断。
(一)核心综合征及眼部表现
- Rieger综合征:常染色体显性遗传。眼部表现为双眼发育缺陷:瞳孔异位、虹膜萎缩、瞳孔洞形成、角膜混浊、虹膜基质发育不全。全身伴牙齿发育不良、面骨畸形,可继发青光眼。
- Alport综合征(眼-耳-肾综合征):主要为X连锁显性遗传。眼部表现为近视、斜视、眼球震颤、圆锥角膜、眼底黄斑区色素沉着。由COL4A5/COL4A3/COL4A4基因突变致病,全身伴感音神经性耳聋和肾小球肾炎。
- 瓦登伯革综合征(耳聋白发眼病综合征):常染色体显性遗传。眼部表现为虹膜异色、内眦外移(眼距宽),全身伴额前白发、听力障碍、鼻根宽阔、并眉。由PAX3、MITF、EDNRB及SOX10等基因突变致病。
- 额鼻发育不良综合征:常染色体显性遗传,分三型。1型有长鼻异常、上睑下垂;2型有无眼畸形、小眼球、低位耳;3型有眼部多部位发育不全。全身伴鼻裂、唇腭裂等面部畸形及骨骼发育异常。
- Apert综合征(尖头并指综合征):常染色体显性遗传。眼部表现为眼距过宽、眼球突出、角膜混浊、斜视。全身伴尖头畸形、面中部发育不良、并指/趾、颅缝早闭。
- Crouzon综合征:常染色体显性遗传。眼部表现为眼距过宽、眼球突出、角膜暴露风险高。全身伴颅面部骨缝早闭、上颌骨发育不良、颅内压增高。
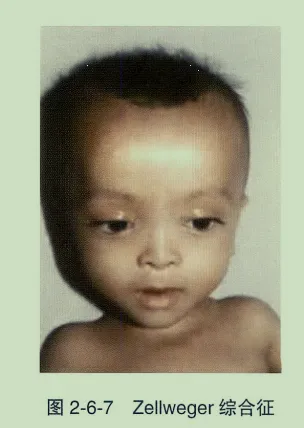
- Zellweger综合征(脑肝肾综合征):常染色体隐性遗传。眼部表现为眼外眦赘皮、晶状体混浊、青光眼、角膜混浊、视网膜发育不全。全身伴面部畸形、肝大、黄疸及神经系统症状。
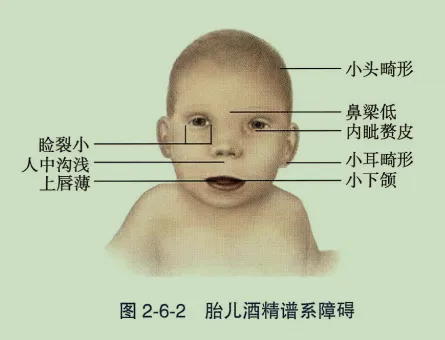
- Noonan综合征:常染色体显性遗传。眼部表现为上睑下垂、眼距宽、虹膜异色、斜视。全身伴特殊面容、先天性心脏病、生长迟缓。
(二)关键致病基因及功能
- PAX6基因(11q13):调控角膜、虹膜、晶状体、视网膜发育。突变可致无虹膜、先天性小眼球、眼前节缺陷、角膜混浊、并发性白内障;纯合突变可致眼球缺失和严重脑缺陷。
- RIEG基因(4q25):调控颅面部和眼部组织发育。突变与Rieger综合征、虹膜缺损、青光眼相关。
- COL4A5/COL4A3/COL4A4基因(X染色体/2号染色体):构成肾小球基底膜和角膜基底膜胶原纤维。突变导致Alport综合征。
- PAX3基因(2q35):调控神经嵴细胞发育,参与虹膜、额部皮肤、听神经发育。突变导致瓦登伯革综合征。
- MITF基因(3p14):调控黑色素细胞发育。突变与瓦登伯革综合征、虹膜异色、小眼球相关。
- SOX10基因(22q13):调控神经嵴细胞分化。突变可致瓦登伯革综合征、小眼球、神经发育异常。
四、先天性眼球震颤
(一)核心定义
指非自主的、有节律性的眼球运动,分为跳动性(眼球向一侧缓慢运动,另一侧快速复位)和摆动性(眼球在两个方向上均做等速摆动)两类,部分患儿可同时存在两种类型。
(二)发病特点
少数婴儿出生时即出现,多数在6月龄内出现。部分类型有遗传性,多为常染色体显性或隐性遗传。随年龄增长,部分患儿眼球震颤可逐渐减轻或消失。
(三)病因分类
- 眼部因素:先天性白内障、先天性青光眼、小眼球、视神经发育不良、视网膜发育不良等,多为单侧或双侧不对称的形觉剥夺性眼球震颤。
- 神经因素:脑干发育异常、小脑病变、颅内感染、颅内肿瘤等,常伴肌张力异常、抽搐、发育迟缓等神经系统症状。
- 遗传因素:基因突变如FRMD7、GPR143等,多为特发性眼球震颤,无明显眼部或神经系统异常。
- 其他因素:早产、缺氧缺血性脑病、胆红素脑病等围生期损伤致神经发育异常。
本文内容整理自相关学习资料,仅为个人学习记录。