读书笔记 | 第02章 学习蛋白质的语言 | 《生物学中的深度学习》
- 2026-05-27 17:41:09

第02章 学习蛋白质的语言
我们所知道的生命活动,本质上都离不开蛋白质(protein)。人类基因组大约包含 20,000 个基因(gene),这些由 DNA 构成的基因,就像一张张蓝图,用来指导不同蛋白质的构建。有些蛋白质的功能比较直接、也比较容易理解,例如胶原蛋白(collagen)为组织提供结构支撑与弹性,血红蛋白(hemoglobin)负责在肺部与身体其他部位之间运输氧气和二氧化碳。另一些蛋白质承担的角色则更抽象一些,它们充当信使、调节器或信号载体,在细胞内部以及细胞之间传递信息。例如,胰岛素(insulin)就是一种蛋白质激素,它会向细胞发出信号,让细胞从血液中吸收糖分。
稍后我们会更详细地讨论 DNA 和蛋白质是如何工作的。但在此之前,你可以先把蛋白质想象成一种鼓鼓囊囊的分子机器,在拥挤的细胞环境中四处碰撞,偶尔发生对生命活动有意义的“有效碰撞”。它的形状和运动看起来也许很混乱,但这两者都经历了数百万年的进化打磨,目的是完成非常具体的分子功能。
本章有一个关键信息点:蛋白质可以表示成一串由基本构件组成的序列,这些构件叫作氨基酸(amino acid)。就像英语使用 26 个字母组成单词一样,蛋白质使用 20 种氨基酸这一“字母表”,拼出具有特定形状和特定任务的长链结构。基于这一点,本章的目标其实很简单:我们要训练一个模型,让它根据蛋白质的氨基酸序列来预测该蛋白质的功能。例如:
给定胶原蛋白 COL1A1的序列(MFSFVDLR...),我们也许会预测它以0.7的概率具有结构性功能,以0.01的概率具有酶功能,等等。给定胰岛素蛋白 INS的序列(MALWMRLL...),我们也许会预测它以0.6的概率具有代谢相关功能,以0.3的概率具有信号传导功能,等等。
提示
如果你想立刻开始动手,最好的方式就是打开配套的 Colab notebook,一边读这一章,一边把代码跑起来。交互式地探索这些例子,是建立直觉、真正把概念记住的最好方式之一。
生物学导引
前面我们已经强调过,蛋白质是细胞内功能执行的核心单位,承担着极其广泛的生物学角色。而蛋白质的功能,与它的三维结构(3D structure)密切相关;这个结构,又是由它的一维氨基酸主序列(primary amino acid sequence)决定的。
如果把信息流再简化回顾一下,那就是:基因编码蛋白质的主氨基酸序列;这条序列决定蛋白质的结构;而结构进一步决定功能。
蛋白质结构
蛋白质结构通常会从四个层级来描述:
一级结构(primary structure):氨基酸的线性序列。 二级结构(secondary structure):局部折叠形成的结构元件,例如 α 螺旋(alpha helix)和 β 折叠(beta sheet)。 三级结构(tertiary structure):整条氨基酸链折叠后形成的整体三维形状。 四级结构(quaternary structure):多个蛋白质亚基(subunit)组装成一个功能复合体的结构层级,当然并不是所有蛋白质都有这一层。
图 2-1 展示了血红蛋白(hemoglobin)在这四个层级上的结构组织方式。
人类遗传密码(genetic code)规定了 20 种主要氨基酸。每一种氨基酸都有独特的化学结构,但它们又可以按一些共有的生化性质进行分组,例如疏水性(hydrophobicity,也就是它们与水相互作用的方式)、电荷(正电、负电或中性),以及极性(polarity,也就是电荷在分子内部的分布是否均匀)。
虽然学过生物化学的人常常都被要求把 20 种氨基酸的名字、结构以及单字母缩写全背下来,但在这里,更实用的做法是优先关注它们在功能上的角色,图 2-2 对这些角色做了一个总结。
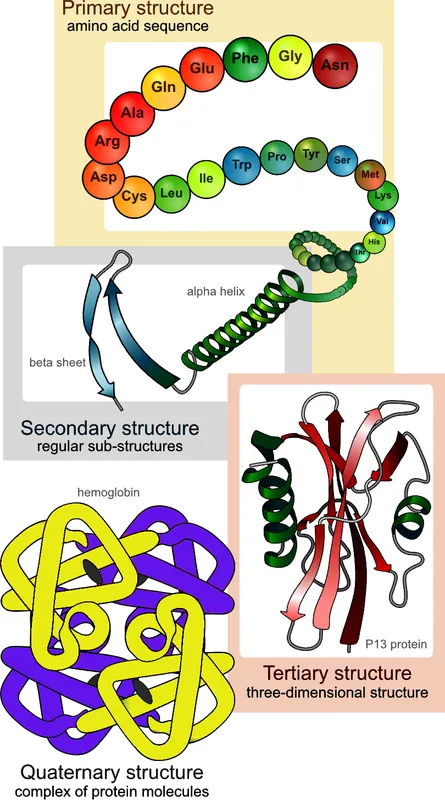
图 2-1. 以血红蛋白为例展示蛋白质结构的四个层级。图片来源:Wikipedia。
例如,D(天冬氨酸,aspartic acid)和 E(谷氨酸,glutamic acid)都带负电,因此它们在很多场景下可以互相替换,而不会显著改变蛋白质功能。但另一些氨基酸承担的角色就要具体得多,哪怕只发生一次单点替换(single substitution),也可能彻底改变蛋白质如何折叠、如何发挥功能,有时甚至会造成严重后果。事实上,许多遗传病就是由这类点突变(point mutation)引起的。一个著名例子是镰刀型贫血(sickle cell anemia):在血红蛋白对应基因中,只发生了一个字母的改变,把一种亲水氨基酸 E 替换成了疏水氨基酸 V,最终导致红细胞形状异常。
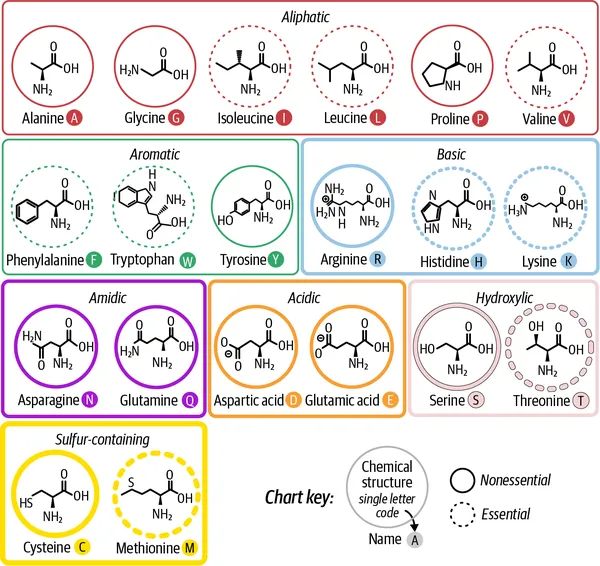
图 2-2. 生物体中 20 种标准氨基酸的化学结构示意图。图中按生化相似性分组,并根据侧链性质(如酸性、碱性、极性、非极性)着色,同时标注了名称、单字母与三字母缩写,以及示例 DNA 密码子(即编码该氨基酸的三联碱基)。改编自 Compound Interest 的信息图。
有了蛋白质结构的基础,我们接下来看看蛋白质的功能,也就是它们在细胞里到底做什么。
蛋白质功能
蛋白质几乎承担了生命活动所需的所有任务:催化化学反应、传递信号、运输分子、提供结构支撑,以及调控基因表达。正因为蛋白质功能如此多样,系统地整理和标注这些功能本身就是一项浩大工程,而其中最常用的框架之一,就是基因本体(Gene Ontology, GO)项目。
GO 系统把蛋白质功能组织为三大类,每一类都刻画了蛋白质在细胞中行为的一个不同侧面:
生物过程(biological process):蛋白质参与的过程,例如细胞分裂、应激响应、碳水化合物代谢或免疫信号传导。 分子功能(molecular function):蛋白质本身执行的具体生化活动,例如与 DNA 结合、与 ATP(细胞内储存并转移能量的分子)结合、充当激酶(kinase,一种把磷酸基团加到其他分子上以改变其活性的酶),或者跨膜运输离子。 细胞组分(cellular component):蛋白质通常位于细胞的哪个位置,例如细胞核、线粒体或细胞外空间。严格来说,这更像一个位置标签,而不是“功能”本身,但它经常能为蛋白质角色提供非常重要的线索,例如位于线粒体中的蛋白质,大概率与能量生产有关。我们会在第 6 章回到这个主题。
同一个蛋白质可以在这些类别上同时拥有多个 GO 注释。例如,一个蛋白质可能既能结合 ATP(分子功能),又参与肌肉收缩(生物过程),同时定位在肌纤维(细胞组分)中。有些注释来自直接实验测定,有些则是通过与已知蛋白的相似性做出的计算推断。本章里,我们会使用一部分经过人工整理、置信度较高、且有实验支持的 GO 注释子集。
预测蛋白质功能
为什么要从蛋白质序列出发预测它的功能?这其实是现代生物学中的一个基础性挑战。下面是几个最常见、也最有影响力的应用场景:
生物技术与蛋白质工程(biotechnology and protein engineering):如果我们能够可靠地从序列预测功能,就有机会进一步设计出具有目标性质的新蛋白质。这对工业化学中的酶设计、医学中的治疗性蛋白,或者合成生物学组件设计都可能非常有用。 疾病机制理解(understanding disease mechanisms):许多疾病都是由特定序列变化,也就是变异(variant)或突变(mutation)破坏蛋白质功能而引起的。一个好的预测模型可以帮助我们判断特定突变会如何改变功能,从而为疾病机制研究和潜在治疗靶点的寻找提供线索。 基因组注释(genome annotation):随着我们不断测序新的物种基因组,越来越多功能未知的蛋白质被发现。对于这些新识别出的蛋白质,尤其是那些与已知蛋白进化关系很远的蛋白质,计算预测是提出功能假设的关键手段。 宏基因组学与微生物组分析(metagenomics and microbiome analysis):当我们测序整个微生物群落时,例如肠道细菌或海洋微生物群,很多编码蛋白质的基因在现有数据库中都没有接近匹配。此时,从序列预测功能能帮助我们揭示这些未知蛋白的角色,从而推进对微生物生态系统及其对宿主或环境影响的理解。
注记
这项任务听起来也许有点像“输入一条序列,输出一个功能”这么直接,但要真正做到准确的蛋白质功能预测,其实非常困难。模型若想成功,必须在隐含层面上理解一整套复杂的生物学规律:氨基酸序列如何决定三维结构,这本身就是一个曾获得诺贝尔奖关注级别的机器学习问题;结构又如何支持功能;而这些功能又如何在动态且极度拥挤的细胞环境中真正发挥作用。
本章不会追求 state-of-the-art 的最强表现。相反,我们的目标是先搭出一个真正能工作的简单模型,并建立一种直觉:蛋白质序列如何被映射到功能注释。沿途我们还会引入一些非常实用的机器学习技巧,例如利用预训练模型提取嵌入(embedding)、把这些嵌入可视化,以及在它们之上训练轻量分类器。这些方法会在后面的章节里反复出现,成为你的常用工具。
机器学习导引
前面我们快速回顾了蛋白质的生物学背景,以及它的功能如何被编码。接下来,我们把注意力转向真正能让机器从蛋白质序列中学习的机器学习技术。
大语言模型
如今几乎走到哪里都会碰到大语言模型(large language model, LLM)。近年来 AI 里许多突破性模型,例如 ChatGPT、Gemini、Claude 和 Llama,都属于这一类。虽然这些模型背后涉及极其庞大的工程体系,但它们最基础的思想其实相当简单:给定前文上下文,去预测下一个 token,例如下一个单词或字符。当然也有一些变体,例如掩码语言模型(masked language model),它会在训练中随机遮掉一些 token,迫使模型学会利用上下文进行推理,但核心原则是一致的。
现代 AI 中一个最令人惊讶的发现是:只要模型足够大,也就是参数量足够多,同时训练数据足够大,也就是 token 总数足够多,那么即使没有显式监督,也会涌现出惊人的能力。这些模型会突然学会摘要、翻译,甚至生成诗歌和故事一类带创造性的内容,尽管训练时根本没有专门把它们当成这些任务来教。
这一点对生物学也很有启发。很多方面上,生物学本身就很像一种“语言”:DNA 和蛋白质都是由离散字母表构成的序列系统,其中存在复杂模式以及强烈依赖上下文的“语法”。如果我们用和自然语言相同的目标,也就是基于上下文预测下一个 token,去在海量生物序列上训练语言模型,那么我们就有机会学到非常丰富的生物学表示。
这些学习得到的表示,可以被用于大量下游任务,例如预测蛋白质功能、推断突变效应,或者识别结构性质,而且不需要每个新任务都从零开始重新训练模型。
本章稍后会介绍目前最成功的蛋白质语言模型之一:ESM2。
嵌入
语言模型最强大、也最通用的输出之一,就是它生成嵌入(embedding)的能力。嵌入本质上是一个数值向量,也就是由浮点数组成的一串数字,用来编码某个实体的意义或结构,这个实体可以是一个词、一个句子,也可以是一条蛋白质序列。例如,一个蛋白质也许会被表示成像 [0.1, -0.3, 1.3, 0.9, 0.2] 这样的向量,它以紧凑数值形式携带了蛋白质的一部分生化或结构信息。
语言模型产生的嵌入并不是随便出来的一堆数字,它们是有组织的:相似输入通常会得到相似嵌入。像 lion、tiger 和 panther 这样的相关单词,会在语言语义空间(semantic space)中聚在一起。同样地,结构或功能相近的蛋白质序列,例如胶原蛋白 I 和胶原蛋白 II,也往往会在一种可以称作“蛋白质空间(protein space)”的地方彼此靠近。
这个思想可以推广到潜在空间(latent space)的概念:那是一个连续而抽象的空间,相似实体会因为学到的模式而被放在彼此靠近的位置。在这种空间中,我们可以做很多强大的操作,例如插值(interpolation)、聚类(clustering)和生成式设计(generative design)。对蛋白质而言,潜在空间甚至能捕捉那些从原始序列本身不容易直接看出来的功能关系。例如,两个蛋白质虽然序列差异很大、进化历史也完全不同,却可能因为趋同演化而承担类似功能,因此在潜在空间里靠得很近。这类表示还可以通过和空间中已经有注释的邻居比较,帮助我们为未知蛋白预测新功能。
注记
如果你想判断蛋白质在结构或功能上是否相似,一个常见做法是使用余弦相似度(cosine similarity)比较它们的嵌入。余弦相似度衡量的是两个向量方向是否一致,而不关心它们的绝对长度。即使两个蛋白质在氨基酸层面差异很大,这种方法仍然能工作。通过计算一个查询蛋白与一组已知蛋白之间的余弦相似度,你可以按嵌入空间中的接近程度对它们排序,而最接近的那些蛋白质,往往会共享功能角色、结构特征或进化背景。
预训练与微调
很多机器学习任务都共享底层结构。无论你的目标是检测仇恨言论、回答法学院入学考试问题,还是写关于水豚(capybara)的诗,一个模型首先都需要掌握语言是如何运作的。因此,我们通常不会为每个任务都从零训练,而是先从一个在庞大、多样化数据集上完成预训练(pretraining)的通用模型出发。
预训练赋予模型广泛知识和一般能力。对于某个具体应用,我们通常会在此基础上再做一次规模更小、目标更聚焦的训练步骤,也就是微调(fine-tuning),让模型在一个领域专门数据集上继续训练。这种“两阶段流程”现在已经是很多机器学习方向里的标准范式,尤其是在预训练语言模型变得越来越强之后。
不过,在本书的第一个技术章节里,我们会采用一种稍有不同的方式。我们并不去微调整个预训练模型,而是把它当作一个冻结的特征提取器(frozen feature extractor):先用它产生嵌入,再在这些嵌入之上训练一个更小的分类器。这个策略计算上更高效,对数据量要求也更低,同时还能充分利用预训练模型已经学到的丰富表示。完整的迁移学习和微调,我们会在后面章节里继续展开。
蛋白质与蛋白质语言模型的表示方式
前面我们已经讨论过蛋白质是什么,也讨论了蛋白质结构如何分层组织:从线性的氨基酸链,到局部折叠,再到真正承载功能的三维形态。为了让这些概念不那么抽象,我们先用 py3Dmol 库加载并可视化一个真实蛋白质结构:
import py3Dmolimport requestsdef fetch_protein_structure(pdb_id: str) -> str: """从 RCSB Protein Data Bank 获取一个 PDB 蛋白质结构。""" url = f"https://files.rcsb.org/download/{pdb_id}.pdb" response = requests.get(url) return response.text# Protein Data Bank(PDB)是蛋白质结构的主要数据库。# 每个结构都有一个唯一的 4 字符 PDB ID。下面给出几个例子。protein_to_pdb = { "insulin": "3I40", # 人类胰岛素:调节葡萄糖摄取。 "collagen": "1BKV", # 人类胶原蛋白:提供结构支撑。 "proteasome": "1YAR", # 古菌蛋白酶体:负责降解蛋白质。}protein = "collagen" # @param ["insulin", "collagen", "proteasome"]pdb_structure = fetch_protein_structure(pdb_id=protein_to_pdb[protein])pdbview = py3Dmol.view(width=400, height=300)pdbview.addModel(pdb_structure, "pdb")pdbview.setStyle({"cartoon": {"color": "spectrum"}})pdbview.zoomTo()pdbview.show()在配套 Colab notebook 中运行这段代码后,你会看到所选蛋白质的交互式三维渲染图。图 2-3 展示的是胶原蛋白(collagen)可视化后的截图。

图 2-3. 使用 py3Dmol 渲染得到的胶原蛋白三维结构。胶原蛋白是一类结构蛋白,会形成三股螺旋纤维,这里可以看到相互缠绕的带状结构。
你也可以试着切换到其他例子,例如 insulin 或 proteasome,感受蛋白质结构的惊人多样性。它们的形状往往直接反映其专门功能。例如,胶原蛋白那种细长而富有弹性的结构,就和它在身体大量组织中充当柔韧支架的作用密切相关。
蛋白质的数值表示
三维可视化对理解很有帮助,但机器学习模型真正需要的是数值输入。因此,若想用机器学习方法分析或建模蛋白质,我们通常会从它的一维氨基酸序列开始。
绝大多数已知生物体的蛋白质序列,都可以从诸如 Uniprot 这样的公共数据库中获取。例如,下面就是人类胰岛素的氨基酸序列:
# 胰岛素前体蛋白序列(后续会被加工成两条蛋白链)。insulin_sequence = ( "MALWMRLLPLLALLALWGPDPAAAFVNQHLCGSHLVEALYLVCGERGFFYTPKTRREAEDLQVGQVELGG" "GPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN")print(f"Length of the insulin protein precursor: {len(insulin_sequence)}.")Output:
Length of the insulin protein precursor: 110.这种序列表示很容易存储、也很容易操作,但在送进机器学习模型之前,它仍然需要先被转换成数值形式。
蛋白质序列的 One-Hot 编码
把蛋白质序列转成数值形式,最简单的方法就是 one-hot encoding。它的基本思路是:
标准氨基酸共有 20 种。 每一种氨基酸用一个长度为 20 的二进制向量表示,其中只有一个位置为 1,表示它是哪一种氨基酸,其他位置全部为0。整条蛋白质序列于是就会被转换成一串 one-hot 向量,也就是序列中每个氨基酸对应一个向量。
下面我们用一个玩具例子来演示:对短蛋白 MALWN 做编码。它正好是胰岛素前体蛋白最前面的五个氨基酸。
首先,我们定义氨基酸字母和整数索引之间的映射关系:
from dlfb.utils.display import print_short_dictamino_acids = [ "R", "H", "K", "D", "E", "S", "T", "N", "Q", "G", "P", "C", "A", "V", "I", "L", "M", "F", "Y", "W",]amino_acid_to_index = { amino_acid: index for index, amino_acid in enumerate(amino_acids)}print_short_dict(amino_acid_to_index)Output:
{'R': 0, 'H': 1, 'K': 2, 'D': 3, 'E': 4, 'S': 5, 'T': 6, 'N': 7, 'Q': 8, 'G': 9}…(+10 more entries)有了这个映射,我们就可以把一条蛋白质序列转成整数序列:
# 甲硫氨酸、丙氨酸、亮氨酸、色氨酸、甲硫氨酸。tiny_protein = ["M", "A", "L", "W", "M"]tiny_protein_indices = [ amino_acid_to_index[amino_acid] for amino_acid in tiny_protein]tiny_protein_indicesOutput:
[16, 12, 15, 19, 16]再进一步,我们就能把这串整数转成 one-hot 编码,如图 2-4 所示。

图 2-4. one-hot 编码会把蛋白质氨基酸序列转成一个二值矩阵,其中每一行对应一个氨基酸位置,每一列对应一种可能残基。矩阵里大多数值都是 0,只有一个位置上的 “1” 表示该处出现了对应氨基酸。
从图 2-4 可以看出:
得到的矩阵形状是 [5, 20],其中 5 行分别对应序列中的 5 个氨基酸位置,而 20 列分别对应 20 种标准氨基酸。每一行除了一个位置是 1之外,其余全部为0。这种表示保留了“类别型实体”的本质,同时不会人为暗示这些氨基酸之间存在数值顺序或大小关系。
注记
为什么不直接跳过 one-hot 这一步,直接把氨基酸整数索引送进模型?
问题在于,数值索引,例如 3 和 17,会暗示出一种人为顺序以及某种“距离关系”,仿佛它们在数值上有可比较性。但氨基酸本质上是类别型实体,并不存在这种有意义的数值关系。
one-hot 编码正是为了避免这种误导:它给每个氨基酸分配一个独立的二值向量,从而确保模型把它们当成彼此平等、彼此分离的类别,而不是从任意索引值中推断出并不存在的模式。
在代码里,我们可以直接使用 JAX 提供的 jax.nn.one_hot 工具来得到这种嵌入:
import jaxone_hot_encoded_sequence = jax.nn.one_hot( x=tiny_protein_indices, num_classes=len(amino_acids))print(one_hot_encoded_sequence)Output:
[[0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 1. 0. 0. 0.] [0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 1. 0. 0. 0. 0. 0. 0. 0.] [0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 1. 0. 0. 0. 0.] [0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 1.] [0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 0. 1. 0. 0. 0.]]我们还可以像图 2-5 那样,把这个 one-hot 编码矩阵画成热图,本质上就是把前面的图 2-4 重新可视化一次:
import seaborn as snsfig = sns.heatmap( one_hot_encoded_sequence, square=True, cbar=False, cmap="inferno")fig.set(xlabel="氨基酸索引", ylabel="蛋白质序列");
图 2-5. 玩具蛋白质序列 MALWM 的 one-hot 编码热图。这个二值矩阵编码了每个残基(residue)的身份,同时不会暗示它们之间存在额外相似性。
现在,我们已经构造出了蛋白质的一个最基础数值表示。接下来,就可以从这种非常朴素的格式继续往前,进入 learned embeddings,也就是能够为每个氨基酸编码更多生物学意义的稠密向量表示(dense vector representation)。
氨基酸的学习型嵌入
在本章剩余部分,我们会使用一个由 Meta 在 2023 年发布的预训练蛋白质语言模型 ESM2(ESM 是 evolutionary scale modeling 的缩写)。这些模型托管在 Hugging Face 平台上。如果你之前还没怎么接触 Hugging Face,那么它是一个非常棒的资源库,里面有成千上万的预训练模型可供直接使用和探索。
稍后我们会更详细地讨论 ESM2 是如何工作的,但在那之前,先来看看它如何表示单个氨基酸。我们会通过 Hugging Face 的 transformers 库来访问它。ESM2 建立在 2017 年提出的 Transformer 神经网络架构之上,而 Transformer 现在已经成为处理文本、蛋白质这类序列数据的标准架构。
注记
理想情况下,我们当然想直接用 JAX / Flax 加载 ESM2,但目前它官方只提供 PyTorch 版本。现实里,熟悉不止一个深度学习框架通常是有帮助的,所以这里我们会先用 PyTorch 加载模型并提取嵌入,然后再用 JAX 在这些嵌入之上继续处理和建模。
后面的章节会重新回到纯 JAX / Flax 工作流,但这里这种“短暂混用框架”的情况,本身就是对真实世界工作流灵活性的一个很好示范。
from transformers import AutoTokenizer, EsmModel# 模型 checkpoint 名称来自这个 GitHub README:# https://github.com/facebookresearch/esm#available-models-and-datasets-model_checkpoint = "facebook/esm2_t33_650M_UR50D"tokenizer = AutoTokenizer.from_pretrained(model_checkpoint)model = EsmModel.from_pretrained(model_checkpoint)我们可以先看看模型内部的 token 到索引映射关系:
vocab_to_index = tokenizer.get_vocab()print_short_dict(vocab_to_index)Output:
{'<cls>': 0, '<pad>': 1, '<eos>': 2, '<unk>': 3, 'L': 4, 'A': 5, 'G': 6, 'V': 7,'S': 8, 'E': 9}…(+23 more entries)这和我们前面手工构造的氨基酸索引很像,但它额外包含了一些特殊 token,例如代表未知残基的 <unk>、表示序列结束的 <eos>,以及比较少见的氨基酸,例如 U(硒代半胱氨酸,selenocysteine)和 O(吡咯赖氨酸,pyrrolysine)。
下面用 ESM2 的 tokenizer 来编码我们那个小型蛋白质序列:
tokenized_tiny_protein = tokenizer("MALWM")["input_ids"]tokenized_tiny_proteinOutput:
[0, 20, 5, 4, 22, 20, 2]如果需要,我们还可以把特殊的起始 token <cls> 和结束 token <eos> 去掉:
tokenized_tiny_protein[1:-1]Output:
[20, 5, 4, 22, 20]接下来,我们通过 model.get_input_embeddings() 从模型里取出学习到的 token 嵌入:
token_embeddings = model.get_input_embeddings().weight.detach().numpy()token_embeddings.shapeOutput:
(33, 1280)这 33 个可能 token 中的每一个,都会被映射到一个 1,280 维空间里。人类当然无法直接可视化这么高维的空间,但我们可以使用诸如 t-SNE 或 UMAP 这类降维方法,把这些嵌入投影到二维平面中,从而以更容易理解的方式观察模型是如何组织不同 token 的:
import pandas as pdfrom sklearn.manifold import TSNEtsne = TSNE(n_components=2, random_state=42)embeddings_tsne = tsne.fit_transform(token_embeddings)embeddings_tsne_df = pd.DataFrame( embeddings_tsne, columns=["first_dim", "second_dim"])embeddings_tsne_df.shapeOutput:
(33, 2)可以看到,经过 t-SNE 处理之后,数组形状变成了 (33, 2),也就是说 33 个 token 现在都被投影到了二维平面上。图 2-6 展示了这些点的散点图,让我们能够直观感受模型是如何组织这些 token 嵌入的。
fig = sns.scatterplot( data=embeddings_tsne_df, x="first_dim", y="second_dim", s=50)fig.set_xlabel("第一维")fig.set_ylabel("第二维");
图 2-6. ESM2 模型所学习 token 嵌入的二维 t-SNE 投影。即便不加任何标签,也已经能隐约看到若干聚类,这暗示模型已经学会以有意义的方式组织这些 token。
为了做一个 sanity check,看看相似类型的 token 是否真的会在二维嵌入空间里聚到一起,我们可以根据已知的氨基酸性质,也就是本章前面提过的那些性质,给每个 token 打上标签,然后重新绘制图 2-7 里的 t-SNE 投影:
from adjustText import adjust_textembeddings_tsne_df["token"] = list(vocab_to_index.keys())token_annotation = { "hydrophobic": ["A", "F", "I", "L", "M", "V", "W", "Y"], "polar uncharged": ["N", "Q", "S", "T"], "negatively charged": ["D", "E"], "positively charged": ["H", "K", "R"], "special amino acid": ["B", "C", "G", "O", "P", "U", "X", "Z"], "special token": [ "-", ".", "<cls>", "<eos>", "<mask>", "<null_1>", "<pad>", "<unk>", ],}embeddings_tsne_df["label"] = embeddings_tsne_df["token"].map( {t: label for label, tokens in token_annotation.items() for t in tokens})fig = sns.scatterplot( data=embeddings_tsne_df, x="first_dim", y="second_dim", hue="label", style="label", s=50,)fig.set_xlabel("第一维")fig.set_ylabel("第二维")texts = [ fig.text(point["first_dim"], point["second_dim"], point["token"]) for _, point in embeddings_tsne_df.iterrows()]adjust_text( texts, expand=(1.5, 1.5), arrowprops=dict(arrowstyle="->", color="grey"));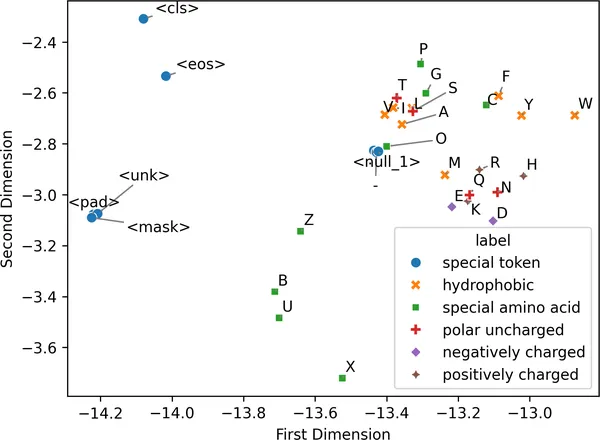
图 2-7. 按氨基酸性质给 t-SNE 投影着色后,可以清楚看到:具有相似生化角色的氨基酸往往会在嵌入空间中形成聚类,这反映出模型确实捕捉到了有意义的生物结构。非氨基酸类的技术 token 也会在这个潜在空间中聚在一起。
具有相似生化性质的 token,确实倾向于彼此聚在一起。例如,疏水氨基酸 F、Y 和 W 会出现在图的右上角,而 <cls> 和 <eos> 这类特殊用途 token,则会一起出现在图左侧。这样的结构说明:模型已经学会根据氨基酸在蛋白质序列中的角色差异,去区分它们。
既然我们已经看过这些 token 嵌入到底长什么样,接下来就该进一步问:ESM2 模型本身到底是如何工作的?它又是怎样学到这些表示的?
ESM2 蛋白质语言模型
现在你已经更熟悉 token embedding 的基本概念了,我们来看看 ESM2 模型到底是怎么工作的。ESM2 是一个掩码语言模型(masked language model, MLM),也就是说,在训练过程中,它会反复随机遮掉每条蛋白质序列中的一部分氨基酸,然后要求模型把这些被遮掉的氨基酸预测出来。对 ESM2 来说,每条序列中大约有随机选中的 15% 氨基酸会在训练时被 mask。图 2-8 对这个过程做了一个直观展示,并把它和自然语言中的掩码语言建模并排比较:

图 2-8. 自然语言模型与蛋白质语言模型中的掩码语言建模对比。在自然语言中,模型会根据上下文去预测缺失的词或子词;蛋白质语言模型则使用完全相同的原则,即随机遮掉序列中的氨基酸,并训练模型根据周围上下文把它们预测出来。
下面我们就在胰岛素蛋白序列里遮掉一个氨基酸,看看模型能不能把它猜出来:
insulin_sequence = ( "MALWMRLLPLLALLALWGPDPAAAFVNQHLCGSHLVEALYLVCGERGFFYTPKTRREAEDLQVGQVELGG" "GPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN")masked_insulin_sequence = ( # 这里把第 29 个位置(从 0 开始计数)的 `L` 氨基酸遮掉: # ...LALLALWGPDPAAAFVNQH L CGSHLVEALYLVCGERGFF... "MALWMRLLPLLALLALWGPDPAAAFVNQH<mask>CGSHLVEALYLVCGERGFFYTPKTRREAEDLQVGQVELGG" "GPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN")# 对加入 mask 的胰岛素序列做 tokenization。masked_inputs = tokenizer(masked_insulin_sequence)["input_ids"]# 检查我们预期的位置上是否真的出现了 <mask> token。注意,# tokenizer 会在序列开头额外加一个 <cls> token,因此这里# 实际上应该在位置 30(而不是 29)看到 <mask>。assert masked_inputs[30] == vocab_to_index["<mask>"]这里的 <mask> token 会告诉模型:请预测这个位置上的氨基酸。为了做到这一点,我们需要加载完整的语言模型 EsmForMaskedLM,因为它包含了用于预测 token 的 language prediction head。
注记
为了加快推理速度,这里我们会换成一个更小的 ESM2 变体,也就是 1.5 亿参数、640 维嵌入的版本,而不再使用前面那个 6.5 亿参数、1,280 维嵌入的大模型。这也提醒我们:Hugging Face 上的很多模型其实都有不同大小的版本,而在它们之间切换,通常只需要换一个 model checkpoint。
当然,这里存在很明确的权衡:更小的模型通常包含的信息更少,在复杂任务上的表现也往往更弱。但它们非常适合快速原型验证和探索模型行为。
下面加载模型:
from transformers import EsmForMaskedLM# 模型 checkpoint 名称来自这个 GitHub README:# https://github.com/facebookresearch/esm#available-models-and-datasets-model_checkpoint = "facebook/esm2_t30_150M_UR50D"tokenizer = AutoTokenizer.from_pretrained(model_checkpoint)masked_lm_model = EsmForMaskedLM.from_pretrained(model_checkpoint)然后运行它来获取对被 mask 掉 token 的预测。图 2-9 展示了结果:模型以非常高的概率正确预测出了 L(亮氨酸,leucine)。
import matplotlib.pyplot as pltmodel_outputs = masked_lm_model( **tokenizer(text=masked_insulin_sequence, return_tensors="pt"))model_preds = model_outputs.logits# 取出 <mask> 位置上的预测结果。mask_preds = model_preds[0, 30].detach().numpy()# 通过 softmax 把模型输出的 logits 转成概率。mask_probs = jax.nn.softmax(mask_preds)# 可视化每个 token 的预测概率。letters = list(vocab_to_index.keys())fig, ax = plt.subplots(figsize=(6, 4))plt.bar(letters, mask_probs, color="grey")plt.xticks(rotation=90)plt.title("被遮盖氨基酸的模型预测概率");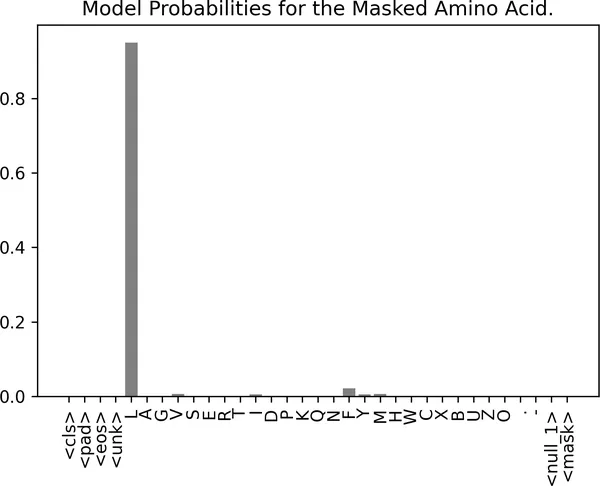
图 2-9. 对胰岛素序列中被遮掉亮氨酸(L)的模型预测。模型以很高概率自信地给出了正确氨基酸 L,这说明它已经学到了蛋白质中的常见序列模式。
我们把这段代码进一步整理成一个更通用的形式 MaskPredictor,让它具备“给序列做 mask”“做预测”“画出预测结果”等方法:
class MaskPredictor: """使用蛋白质语言模型预测被遮盖的氨基酸。""" def __init__(self, tokenizer: PreTrainedTokenizer, model: PreTrainedModel): """使用 tokenizer 和预训练模型完成初始化。""" self.tokenizer = tokenizer self.model = model def plot_predictions(self, sequence: str, mask_index: int) -> Figure: """绘制被遮盖氨基酸的预测概率图。""" mask_probs = self.predict(sequence, mask_index) fig, _ = plt.subplots(figsize=(6, 4)) plt.bar(list(self.tokenizer.get_vocab().keys()), mask_probs, color="grey") plt.xticks(rotation=90) plt.title( "被遮盖氨基酸的模型预测概率\n" f"位置={mask_index}(真实氨基酸 = {sequence[mask_index]})" ) return fig def predict(self, sequence: str, mask_index: int) -> jax.Array: """返回某个位置被遮盖氨基酸的模型预测概率。""" masked_sequence = self.mask_sequence(sequence, mask_index) masked_inputs = self.tokenizer(masked_sequence, return_tensors="pt") model_outputs = self.model(**masked_inputs) mask_preds = model_outputs.logits[0, mask_index + 1].detach().numpy() mask_probs = jax.nn.softmax(mask_preds) return mask_probs @staticmethod def mask_sequence(sequence: str, mask_index: int) -> str: """在输入序列的指定位置插入 mask token。""" if mask_index < 0 or mask_index > len(sequence): raise ValueError("Mask 索引超出了序列范围。") return f"{sequence[0:mask_index]}<mask>{sequence[(mask_index + 1):]}"现在换一个位置试试看,也就是索引 26 处,这里的真实氨基酸是 N(天冬酰胺,asparagine)。结果见图 2-10:
MaskPredictor(tokenizer, model=masked_lm_model).plot_predictions( sequence=insulin_sequence, mask_index=26);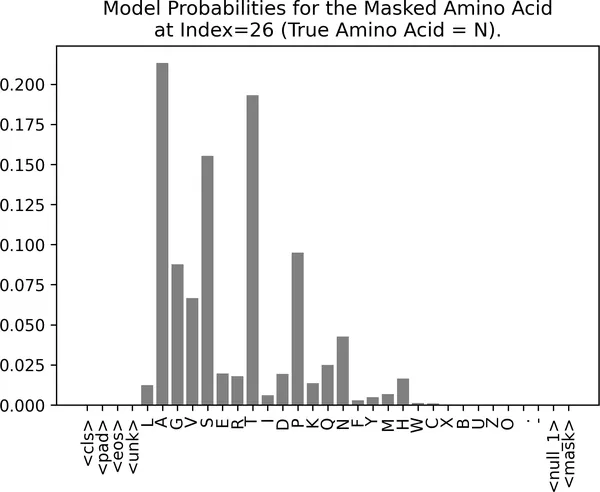
图 2-10. 对胰岛素序列中被遮掉天冬酰胺(N)的模型预测。这里模型明显更不确定,它给多个候选氨基酸都分配了中等概率,这说明这个位置仅依靠周围上下文更难被准确预测。
在这个例子里,模型并没有对某一个氨基酸表现出特别强的偏好。它给了多个候选中等概率,例如 A、T 和 S,而真实氨基酸 N 的概率反而并不高。这种不确定性,可能反映了该位置在生化层面上的灵活性:蛋白质的某些区域由于冗余、结构弹性,或者并没有那么严格的功能约束,因此能够容忍不同残基(residue)出现。人们通常把这种位置称为“permissive positions”,它们在无序区域(disordered / unstructured region)或蛋白质表面区域中尤其常见。
这个例子说明,模型确实已经学会了蛋白质的概率语法(probabilistic grammar)。接下来的问题就是:我们如何把这种理解进一步用于表示“整个蛋白质”,而不是一次只盯着一个氨基酸?
提取整条蛋白质嵌入的策略
到目前为止,我们关注的还是 ESM2 如何表示单个氨基酸。但很多下游任务,例如蛋白质功能预测,需要的是“整条蛋白质序列”的固定长度表示。那么,我们该如何把一条变长的氨基酸序列,压缩成一个同时携带整体结构和意义的 embedding 向量呢?
常见做法主要有几种:
把氨基酸嵌入拼接起来。一个直接方法是遍历序列中的每个氨基酸,取出它的 embedding,然后把它们首尾拼接成一个超长向量。例如,如果某个蛋白长度是 10,每个氨基酸 embedding 维度是640,那么最终就会得到一个长度10 × 640 = 6400的蛋白质 embedding。这种方法确实保留了每个氨基酸的细粒度信息,但问题也很明显:长度不固定。不同蛋白质会产生不同长度的 embedding,给模型输入格式带来麻烦。 可扩展性差。长蛋白质会产出巨大的向量。例如 titin 作为已知最长的人类蛋白,大约有 34,000 个氨基酸,按这种方式会生成超过 4,300 万个数值,这对大多数模型来说都过于笨重。 建模能力有限。这种做法把每个氨基酸几乎当成独立实体看待,忽略了对蛋白质功能至关重要的上下文关系。 对氨基酸嵌入求平均。一个更紧凑的做法,是对整条序列里的 token embedding 取平均。仍然以长度 10、每个 embedding 为640维的蛋白质为例,我们可以把 10 个 embedding 在序列维度上求平均,得到一个最终 640 维向量。这样做的优点是,无论蛋白质长短,输出向量维度都固定,效率也不错,因此实践中确实常用。但它也很粗糙,因为平均会抹掉顺序信息和相互作用信息。你可以把它想象成:把一本小说里所有单词向量求平均,确实还能留下些大意,但细节和层次感会丢掉很多。使用模型的上下文化序列嵌入(contextual sequence embeddings)。更有原则的做法,是直接从语言模型里提取整条序列的隐藏表示(hidden representation)。因为 ESM2是通过“根据上下文预测被遮住 token”的目标训练出来的,所以它内部各层为序列中每个氨基酸编码的,都是富含上下文信息的表示。更具体地说,我们可以把整条蛋白质序列送进ESM2,提取最后一层隐藏状态,从而得到一个形状为(L', D)的张量,其中L'是输出 token 数量,它有时会和输入长度L略有不同,而D是模型隐藏维度,例如640。随后,我们再沿序列长度维度做 mean pooling,得到一个固定长度、形状为(D,)的向量。虽然平均听起来还是有点“粗暴”,但在实践中它经常 surprisingly effective,因为模型已经通过 self-attention 把上下文信息融进了每个 token 的表示里,因此池化后的结果依然保留了跨整个序列的重要依赖关系。
最后这一种,也是实践中最常见、通常也最强的做法,而下一节我们就会具体使用它。
细胞外蛋白与膜蛋白的嵌入
关于 GO 数据集,我们会在下一节真正开始做蛋白质功能预测时再系统介绍。这里先提前借用它,把每个 UniProt 蛋白 accession 和序列,与它已知的细胞定位(cellular location)关联起来:
import pandas as pdfrom dlfb.utils.context import assetsprotein_df = pd.read_csv(assets("proteins/datasets/sequence_df_cco.csv"))protein_df = protein_df[~protein_df["term"].isin(["GO:0005575", "GO:0110165"])]num_proteins = protein_df["EntryID"].nunique()print(protein_df)Output:
EntryID Sequence taxonomyID term aspect Length0 O95231 MRLSSSPPRGPQQLSS... 9606 GO:0005622 CCO 2581 O95231 MRLSSSPPRGPQQLSS... 9606 GO:0031981 CCO 2582 O95231 MRLSSSPPRGPQQLSS... 9606 GO:0043229 CCO 258... ... ... ... ... ... ...337551 E7ER32 MPPLKSPAAFHEQRRS... 9606 GO:0031974 CCO 798337552 E7ER32 MPPLKSPAAFHEQRRS... 9606 GO:0005634 CCO 798337553 E7ER32 MPPLKSPAAFHEQRRS... 9606 GO:0005654 CCO 798[294731 rows x 6 columns]对于由 EntryID 标识的每条蛋白质序列,term 列给出了它在细胞定位上的 GO 注释。
这里我们先聚焦两个具体位置:
extracellular (GO:0005576):分泌到细胞外的蛋白质,常常与信号传导、免疫反应或结构性作用相关。 membrane (GO:0016020):嵌入细胞膜或与细胞膜相关联的蛋白质,通常承担运输、信号传导或细胞-细胞相互作用等功能。
细胞位置与序列特征之间的联系
这里我们是按“细胞定位”来筛选蛋白质,但手头这个模型明明只是在序列上训练出来的,那么它们之间到底有什么联系? 核心点在于:某些类型的蛋白质往往具有非常有代表性的序列特征,而这些特征又和它们在细胞中的工作位置高度相关。例如,膜蛋白(membrane protein)常常包含一段段能够把自己锚定在细胞膜中的氨基酸区域。这些区域通常比较疏水(hydrophobic),从而更容易与富含脂质的膜环境相互作用。 相比之下,细胞外蛋白(extracellular protein)往往带有短的信号序列(signal sequence),用于引导它们被分泌出去。它们也经常通过二硫键(disulfide bond)形成更稳定的结构,并且可能包含帮助它们与其他分子结合的区域。 这些结构性特征都被编码在氨基酸序列之中。理论上,即便 ESM2 这样的预训练语言模型从未见过“位置标签”,它也应该能够从序列里学到这些信号。本节其实就是在测试:这些结构信号,是否已经被反映在模型学到的 embedding 中。
接下来,我们把数据集过滤到只保留“只属于这两个位置之一”的蛋白质:
# 把蛋白质数据框过滤到只保留单一定位的蛋白质。num_locations = protein_df.groupby("EntryID")["term"].nunique()proteins_one_location = num_locations[num_locations == 1].indexprotein_df = protein_df[protein_df["EntryID"].isin(proteins_one_location)]go_function_examples = { "extracellular": "GO:0005576", "membrane": "GO:0016020",}sequences_by_function = {}min_length = 100max_length = 500 # 限制序列长度,以节省时间和内存。num_samples = 20for function, go_term in go_function_examples.items(): proteins_with_function = protein_df[ (protein_df["term"] == go_term) & (protein_df["Length"] >= min_length) & (protein_df["Length"] <= max_length) ] print( f"找到 {len(proteins_with_function)} 个人人类蛋白质\n" f"其对应定位类别为 '{function}'({go_term}),\n" f"并且满足 {min_length}<=length<={max_length}。\n" f"现随机抽样 {num_samples} 条蛋白质序列。\n" ) sequences = list( proteins_with_function.sample(num_samples, random_state=42)["Sequence"] ) sequences_by_function[function] = sequencesOutput:
找到 164 个人人类蛋白质其对应定位类别为 'extracellular'(GO:0005576),并且满足 100<=length<=500。现随机抽样 20 条蛋白质序列。找到 65 个人人类蛋白质其对应定位类别为 'membrane'(GO:0016020),并且满足 100<=length<=500。现随机抽样 20 条蛋白质序列。接下来,我们从这些序列中提取 embedding。函数 get_mean_embeddings 会计算每条序列沿长度维度的隐藏状态均值,也就是把模型对整条蛋白质序列的表示做一个汇总:
def get_mean_embeddings( sequences: list[str], tokenizer: PreTrainedTokenizer, model: PreTrainedModel, device: torch.device | None = None,) -> np.ndarray: """Compute mean embedding for each sequence using a protein LM.""" if not device: device = get_device() # 对输入序列做 tokenization,并补齐到相同长度。 model_inputs = tokenizer(sequences, padding=True, return_tensors="pt") # 把 tokenized 输入移动到目标设备(CPU 或 GPU)上。 model_inputs = {k: v.to(device) for k, v in model_inputs.items()} # 把模型移动到目标设备,并切换到评估模式。 model = model.to(device) model.eval() # 在不跟踪梯度的情况下执行前向传播,得到嵌入。 with torch.no_grad(): outputs = model(**model_inputs) mean_embeddings = outputs.last_hidden_state.mean(dim=1) return mean_embeddings.detach().cpu().numpy()这里我们使用一个更小的 ESM2 模型来提取 embedding。它输出的是 320 维表示,而且比更大的变体明显更省内存:
model_checkpoint = "facebook/esm2_t6_8M_UR50D"tokenizer = AutoTokenizer.from_pretrained(model_checkpoint)model = EsmModel.from_pretrained(model_checkpoint)然后开始实际计算 embedding:
# 为每一种定位计算平均蛋白质嵌入。protein_embeddings = { loc: get_mean_embeddings(sequences_by_function[loc], tokenizer, model) for loc in ["extracellular", "membrane"]}# 重新整理数据格式。labels, embeddings = [], []for location, embedding in protein_embeddings.items(): labels.extend([location] * embedding.shape[0]) embeddings.append(embedding) print(f"{location}: {embedding.shape}")Output:
extracellular: (20, 320)membrane: (20, 320)现在,每一组 20 条采样蛋白都被表示成了一个 (20, 320) 的 embedding 矩阵。这意味着:对每一条序列来说,无论它原本有多长,我们最终都得到了一个固定长度、320 维的向量。这些向量对应于“最后一层隐藏状态在序列维度上求平均”的结果,因此理应携带关于蛋白质整体结构的一部分信息。
为了看这些 embedding 是否真的和蛋白质定位有关,我们再用 t-SNE 把它们投影到二维平面中。这是一种非常常见的高维数据可视化方法。图 2-11 展示的结果表明:细胞外蛋白和膜蛋白在这个空间中,确实倾向于形成彼此不同的聚类。
import numpy as npimport seaborn as snsfrom sklearn.manifold import TSNEembeddings_tsne = TSNE(n_components=2, random_state=42).fit_transform( np.vstack(embeddings))embeddings_tsne_df = pd.DataFrame( { "first_dimension": embeddings_tsne[:, 0], "second_dimension": embeddings_tsne[:, 1], "location": np.array(labels), })fig = sns.scatterplot( data=embeddings_tsne_df, x="first_dimension", y="second_dimension", hue="location", style="location", s=50, alpha=0.7,)plt.title("tSNE of Protein Embeddings")plt.title("蛋白质嵌入的 t-SNE 投影")fig.set_xlabel("第一维")fig.set_ylabel("第二维");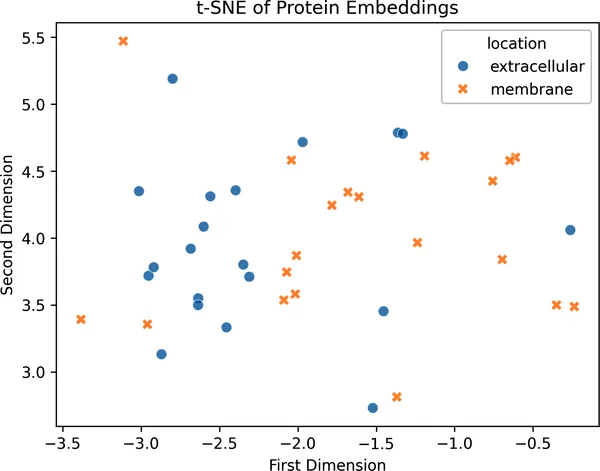
图 2-11. 来自一个小型 ESM2 模型的 320 维 embedding 的二维 t-SNE 投影。即便是这个轻量模型,我们也能看到细胞外蛋白和膜蛋白倾向于形成不同聚类,这说明 embedding 中确实包含与细胞定位相关的信息。
虽然这种分离并不完美,但趋势已经很清楚:细胞外蛋白和膜蛋白,确实会在 embedding 空间里倾向于出现在不同区域。更令人惊讶的是,模型完全是单靠序列就捕捉到了这一点。这说明:即便没有任何显式的细胞定位监督信号,学习到的 embedding 依然反映了有生物学意义的模式。
有了这个初步探索,我们现在终于转向本章的核心机器学习任务:预测蛋白质功能。先从准备数据集开始。
准备数据
很多机器学习图书和博客文章,都会尽快跳到最“刺激”的部分,也就是训练模型和评估模型。但在真实工作流里,训练往往只占整个流程中很小的一部分。大量时间其实花在理解数据、清洗数据和组织数据上。而且,当模型出现问题时,根因往往也藏在数据里。因此,我们不会像魔术一样直接把一份打磨好的 CSV 扔到你面前,而是会一步一步带你走完整个数据准备过程:从真实世界资源出发,再一步步把它们处理成模型真正可用的形式。
我们的目标,是让模型能够从序列预测蛋白质功能,这意味着我们需要组装出一个由 (protein_sequence, protein_function) 对构成的数据集。幸运的是,生物学界早已发展出比较系统的蛋白质功能定义框架,同时也已经积累了一些人工整理的数据集。其中最常用的资源之一,就是 CAFA(Critical Assessment of Functional Annotation)挑战赛。这是一个社区驱动的竞赛,参赛团队会构建模型来预测蛋白质功能。我们会把 CAFA 数据作为原材料,但后续的处理、过滤和结构化工作,仍然需要我们自己来完成。
注记
如果你熟悉 AlphaFold 和蛋白质结构预测,那么你可能也听过名字很像的 CASP(Critical Assessment of Structure Prediction)。它在蛋白质结构预测社区中扮演着和 CAFA 类似的角色。像这样公开、可复用的 benchmark,对推动计算生物学很多问题上的进展都起到了非常关键的作用。
现在,我们就开始进入 CAFA 数据集本身。
加载 CAFA3 数据
CAFA 挑战已经举办过好几轮,但 CAFA3 是目前最新一个公开可用的数据集。我们首先从 CAFA 官网下载了 “CAFA3 Targets” 和 “CAFA3 Training Data” 这两组文件。现在先从标签文件开始加载,它记录了每个蛋白质对应的功能注释:
labels = pd.read_csv( assets("proteins/datasets/train_terms.tsv.zip"), sep="\t", compression="infer")print(labels)Output:
EntryID term aspect0 A0A009IHW8 GO:0008152 BPO1 A0A009IHW8 GO:0034655 BPO2 A0A009IHW8 GO:0072523 BPO... ... ... ...5363860 X5M5N0 GO:0005515 MFO5363861 X5M5N0 GO:0005488 MFO5363862 X5M5N0 GO:0003674 MFO[5363863 rows x 3 columns]这个数据框包含三列:
EntryID:蛋白质的 UniProt ID。 term:描述某个具体蛋白质功能的 GO accession code。 aspect:这个功能所属的 GO 大类,也就是前文介绍过的三类之一:生物过程(BPO)、分子功能(MFO)和细胞组分(CCO)。
term 这一列里目前只有 GO 编号。为了让它更容易理解,我们最好能拿到这些编号对应的人类可读描述。这个信息在 CAFA 文件里并不直接提供,但可以从 Gene Ontology 的下载页面获取。GO 本体以 .obo 文件的图结构格式存储,我们可以用 Python 库 obonet 来解析它。下面就是获取这些 term 描述的方法:
import obonetdef get_go_term_descriptions(store_path: str) -> pd.DataFrame: """返回 GO term 到描述文本的映射;若本地不存在则先下载。""" if not os.path.exists(store_path): url = "https://current.geneontology.org/ontology/go-basic.obo" graph = obonet.read_obo(url) # 从图节点中提取 GO term 的 ID 和名称。 id_to_name = {id: data.get("name") for id, data in graph.nodes(data=True)} go_term_descriptions = pd.DataFrame( zip(id_to_name.keys(), id_to_name.values()), columns=["term", "description"], ) go_term_descriptions.to_csv(store_path, index=False) else: go_term_descriptions = pd.read_csv(store_path) return go_term_descriptions如果本地已经有这个注释文件,函数就直接读取;如果没有,就先下载并缓存:
go_term_descriptions = get_go_term_descriptions( store_path=assets("proteins/datasets/go_term_descriptions.csv"))print(go_term_descriptions)Output:
term description0 GO:0000001 mitochondrion in...1 GO:0000002 mitochondrial ge...2 GO:0000006 high-affinity zi...... ... ...40211 GO:2001315 UDP-4-deoxy-4-fo...40212 GO:2001316 kojic acid metab...40213 GO:2001317 kojic acid biosy...[40214 rows x 2 columns]然后,我们把这些人类可读的 term 描述合并回 labels 数据框:
labels = labels.merge(go_term_descriptions, on="term")labelsOutput:
EntryID term aspect description0 A0A009IHW8 GO:0008152 BPO metabolic process1 A0A009IHW8 GO:0034655 BPO nucleobase-conta...2 A0A009IHW8 GO:0072523 BPO purine-containin...... ... ... ... ...4933955 X5M5N0 GO:0005515 MFO protein binding4933956 X5M5N0 GO:0005488 MFO binding4933957 X5M5N0 GO:0003674 MFO molecular_function[4933958 rows x 4 columns]在本章里,我们只聚焦分子功能(MFO),也就是蛋白质在生化层面究竟“做了什么”。后面如果你愿意,也完全可以把本章的方法扩展到另外两类 GO 注释上。
先看看这个数据集中最常见的分子功能标签有哪些:
labels = labels[labels["aspect"] == "MFO"]print(labels["description"].value_counts())Output:
descriptionmolecular_function 78637binding 57380protein binding 47987 ... kaempferide 7-O-methyltransferase activity 1protopine 6-monooxygenase activity 1costunolide 3beta-hydroxylase activity 1Name: count, Length: 6973, dtype: int64我们已经能看出,功能标签的分布极度偏斜。有些 term,例如 molecular_function、binding 和 protein binding,会出现数万次;而另一些 term 可能只出现一次。像 molecular_function 这种标签其实过于泛化,几乎不提供什么真正有区分度的信息,因此对机器学习帮助不大。后面我们会把它们过滤掉。
接下来,加载与每个蛋白质 ID 对应的蛋白质序列。这部分信息存放在 train_sequences.fasta 文件中。.fasta 是生物学里表示蛋白质序列或 DNA 序列的标准格式之一。我们可以借助 BioPython 的 SeqIO 模块,把这个 .fasta 文件解析成更容易处理的格式。
注记
顺便说一句:没有人一开始就知道 BioPython 的 SeqIO 模块是什么、.fasta 文件怎么用,或者 GO 注释到底代表什么,这完全正常。做生物学和机器学习交叉领域的工作,本来就意味着你会不断接触新工具和新术语。需要频繁去查新概念,不只是可以接受,而是很正常的一部分。
我们把 .fasta 序列转换成一个 pandas 数据框,这样后续处理会容易很多:
from Bio import SeqIOsequences_file = assets("proteins/datasets/train_sequences.fasta")fasta_sequences = SeqIO.parse(open(sequences_file), "fasta")data = []for fasta in fasta_sequences: data.append( { "EntryID": fasta.id, "Sequence": str(fasta.seq), "Length": len(fasta.seq), } )sequence_df = pd.DataFrame(data)print(sequence_df)Output:
EntryID Sequence Length0 P20536 MNSVTVSHAPYTITYH... 2181 O73864 MTEYRNFLLLFITSLS... 3542 O95231 MRLSSSPPRGPQQLSS... 258... ... ... ...142243 Q5RGB0 MADKGPILTSVIIFYL... 448142244 A0A2R8QMZ5 MGRKKIQITRIMDERN... 459142245 A0A8I6GHU0 HCISSLKLTAFFKRSF... 138[142246 rows x 3 columns]我们还顺手计算了每条序列的长度,因为蛋白质长度差异可能非常大,而这个信息在后续过滤数据时会很有用。
这里还有一个很重要的细节:CAFA 数据集包含来自很多不同物种的蛋白质。为了只保留人类蛋白,我们要用下载包里附带的 taxonomy 文件:
taxonomy_file = assets("proteins/datasets/train_taxonomy.tsv.zip")taxonomy = pd.read_csv(taxonomy_file, sep="\t", compression="infer")print(taxonomy)Output:
EntryID taxonomyID0 Q8IXT2 96061 Q04418 5592922 A8DYA3 7227... ... ...142243 A0A2R8QBB1 7955142244 P0CT72 284812142245 Q9NZ43 9606[142246 rows x 2 columns]这个文件给每条蛋白质都附带了一个 taxonomy ID(taxonomyID),它来自 NCBI 的物种分类系统。我们把它合并进序列数据框,并只保留 taxonomyID == 9606 的记录,也就是 Homo sapiens(人类):
sequence_df = sequence_df.merge(taxonomy, on="EntryID")sequence_df = sequence_df[sequence_df["taxonomyID"] == 9606]现在来概览一下,经过过滤后的数据集里有多少个唯一蛋白质,以及多少种分子功能:
sequence_df = sequence_df.merge(labels, on="EntryID")print( f'Dataset contains {sequence_df["EntryID"].nunique()} human proteins ' f'with {sequence_df["term"].nunique()} molecular functions.')Output:
Dataset contains 16336 human proteins with 4101 molecular functions.再来看一下把功能标签合并进去之后的 sequence_df 长什么样:
print(sequence_df)Output:
EntryID Sequence Length taxonomyID term aspect \0 O95231 MRLSSSPPRGPQQLSS... 258 9606 GO:0003676 MFO 1 O95231 MRLSSSPPRGPQQLSS... 258 9606 GO:1990837 MFO 2 O95231 MRLSSSPPRGPQQLSS... 258 9606 GO:0001216 MFO ... ... ... ... ... ... ... 152523 Q86TI6 MGAAAVRWHLCVLLAL... 347 9606 GO:0005515 MFO 152524 Q86TI6 MGAAAVRWHLCVLLAL... 347 9606 GO:0005488 MFO 152525 Q86TI6 MGAAAVRWHLCVLLAL... 347 9606 GO:0003674 MFO description 0 nucleic acid bin... 1 sequence-specifi... 2 DNA-binding tran... ... ... 152523 protein binding 152524 binding 152525 molecular_function [152526 rows x 7 columns]从这个表里已经能看出,很多蛋白质都同时关联了多个分子功能。为了更系统地量化这一点,我们来看图 2-12 中“每个蛋白质对应多少个功能”的分布:
sequence_df.groupby("EntryID")["term"].nunique().plot.hist( bins=100, figsize=(5, 3), color="grey", log=True)plt.xlabel("Number of Molecular Function Annotations per Protein")plt.ylabel("Frequency (log scale)")plt.title("Distribution of Function Counts per Protein")plt.tight_layout()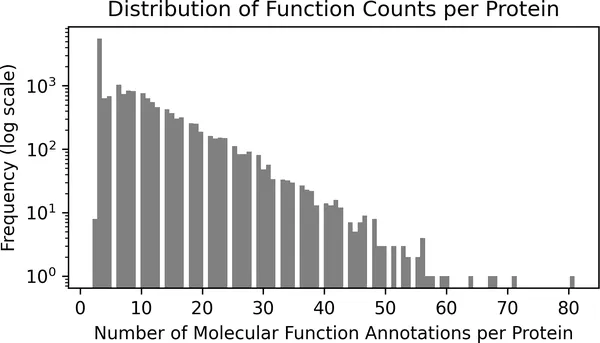
图 2-12. 每个蛋白质对应分子功能注释数量的分布。为了让稀有情况更容易看见,纵轴使用了对数刻度。大多数蛋白质的注释功能少于 20 个,但也有少数蛋白质会关联超过 50 个不同的分子角色。
这种模式反映了真实生物学中的复杂性:很多蛋白质只执行一个定义明确的功能,但另一些蛋白质会参与多种分子层面的角色。例如,有些蛋白既是酶、又会与其他分子结合,还会参与多个通路。从机器学习的角度看,这意味着我们的模型必须能给同一个蛋白质同时打上多个功能标签,而且还要应对“有些标签非常常见、有些标签非常稀有”这一现实。
现在我们进一步仔细看看最常见的分子功能标签。有些 term 过于宽泛,几乎所有蛋白质都会被分到这类标签里,因此没有太大区分价值。比如,molecular function 几乎适用于所有蛋白质,binding 覆盖了 93%,protein binding 出现在 89% 的样本里。训练时,这些标签会在损失函数里占据主导地位,导致模型过度沉迷于预测它们,而忽略更有意义的功能。因此,作为一个数据预处理步骤,我们先显式去掉这些过于泛化的 term:
uninteresting_functions = [ "GO:0003674", # “molecular function”,适用于 100% 的蛋白质。 "GO:0005488", # “binding”,适用于 93% 的蛋白质。 "GO:0005515", # “protein binding”,适用于 89% 的蛋白质。]sequence_df = sequence_df[~sequence_df["term"].isin(uninteresting_functions)]sequence_df.shapeOutput:
(106501, 7)在分布的另一端,也有一些分子功能极其稀有。比如 GO:0099609(microtubule lateral binding)只出现了一次。要让模型学到有意义的对应关系,每种功能至少需要有足够的训练样本。因此,我们还会过滤掉最稀有的标签,只保留那些至少出现在 50 个蛋白质中的功能:
common_functions = ( sequence_df["term"] .value_counts()[sequence_df["term"].value_counts() >= 50] .index)sequence_df = sequence_df[sequence_df["term"].isin(common_functions)]sequence_df["term"].value_counts()Output:
termGO:0003824 3875GO:1901363 2943GO:0003676 2469 ... GO:0031490 51GO:0019003 50GO:0015179 50Name: count, Length: 303, dtype: int64这样就得到了一组更干净、也更适合学习的功能标签集合。
注记
数据处理过程中用到的阈值,例如“某个标签至少出现多少次才保留”,在一定程度上都带有任意性,但它们会显著影响模型表现。这类决定本质上就是超参数(hyperparameter),应该结合具体任务、数据集规模以及模型容量来调节。
现在,我们把数据框重塑一下:让每一行对应一个蛋白质,每一列对应一个分子功能标签。我们会使用 pandas 的 pivot 函数,把数据变成这种多标签格式:
sequence_df = ( sequence_df[["EntryID", "Sequence", "Length", "term"]] .assign(value=1) .pivot( index=["EntryID", "Sequence", "Length"], columns="term", values="value" ) .fillna(0) .astype(int) .reset_index())print(sequence_df)Output:
term EntryID Sequence Length GO:0000166 GO:0000287 ... \0 A0A024R6B2 MIASCLCYLLLPATRL... 670 0 0 ... 1 A0A087WUI6 MSRKISKESKKVNISS... 698 0 0 ... 2 A0A087X1C5 MGLEALVPLAMIVAIF... 515 0 0 ... ... ... ... ... ... ... ... 10706 Q9Y6Z7 MNGFASLLRRNQFILL... 277 0 0 ... 10707 X5D778 MPKGGCPKAPQQEELP... 421 0 0 ... 10708 X5D7E3 MLDLTSRGQVGTSRRM... 237 0 0 ... term GO:1901702 GO:1901981 GO:1902936 GO:1990782 GO:1990837 0 0 0 0 0 0 1 0 0 0 0 0 2 0 0 0 0 0 ... ... ... ... ... ... 10706 0 0 0 0 0 10707 0 0 0 0 0 10708 0 0 0 0 0 [10709 rows x 306 columns]很好,现在这个数据集已经几乎是可以直接送进机器学习流程的格式了。在继续往前之前,我们先做几个最后的 sanity check。
首先,看看现在总共有多少个唯一蛋白质:
sequence_df["EntryID"].nunique()Output:
10709这个数量大致合理。人类基因组里大约有 21,000 个蛋白质编码基因,而我们前面又做了多轮过滤,所以最后得到一个略小一些的数字是很正常的。做这类工作时,始终保留一个量级上的直觉非常重要。如果这里出现的是 1,000 或 1,000,000,我们就应该立刻怀疑哪里出了问题。
接着,检查一下是否存在重复的蛋白质序列:
sequence_df["Sequence"].nunique()Output:
10698看起来确实有少量蛋白质序列重复。例如,P0DP23、P0DP24 和 P0DP25 这几个 entry 共用同一条序列:
print(sequence_df[sequence_df["EntryID"].isin(["P0DP23", "P0DP24", "P0DP25"])])Output:
term EntryID Sequence Length GO:0000166 GO:0000287 ... \1945 P0DP23 MADQLTEEQIAEFKEA... 149 0 0 ... 1946 P0DP24 MADQLTEEQIAEFKEA... 149 0 0 ... 1947 P0DP25 MADQLTEEQIAEFKEA... 149 0 0 ... term GO:1901702 GO:1901981 GO:1902936 GO:1990782 GO:1990837 1945 0 0 0 0 0 1946 0 0 0 0 0 1947 0 0 0 0 0 [3 rows x 306 columns]这些看起来属于真实存在的生物学重复,即不同 UniProt 标识符对应了完全相同的蛋白质序列,因此我们保留它们。
到这一步,我们已经得到了一个最终数据集:它把 10,709 个人人类蛋白质,与 303 种分子功能中的一个或多个关联起来。
不过,由于我们这里使用的“平均嵌入”方案在内存上还是比较吃紧,所以再做一个额外过滤:只保留长度不超过 500 个氨基酸的蛋白质。这样可以减少模型推理和训练时出现内存溢出的风险:
print(sequence_df.shape)sequence_df = sequence_df[sequence_df["Length"] <= 500]print(sequence_df.shape)Output:
(10709, 306)(5957, 306)这个步骤大约把数据集减半,但对于初始原型验证来说完全没问题。如果后面时间和内存都允许,你随时可以去掉这个限制。
现在,我们已经有了一份干净而紧凑的数据集,接下来继续把它处理成更适合机器学习使用的形式。
将数据集划分为不同子集
我们会把数据集拆成三个互不重叠的子集:
训练集(training set):用于拟合模型。模型在训练过程中会看到这部分数据,并据此学习模式。 验证集(validation set):用于在开发阶段评估模型表现。我们会用它来调超参数,并比较不同模型变体。 测试集(test set):只在最后做一次最终评估时使用。最关键的是,我们不能用它来指导模型设计决策。它是我们估计模型面对真正未见数据时泛化能力的最佳依据。
我们按照蛋白质的 EntryID 来划分,确保每个蛋白只会出现在一个子集中:
from sklearn.model_selection import train_test_split# 60% 的蛋白质进入训练集。train_sequence_ids, valid_test_sequence_ids = train_test_split( list(set(sequence_df["EntryID"])), test_size=0.40, random_state=42)# 剩余的 40% 再平均拆成验证集和测试集。valid_sequence_ids, test_sequence_ids = train_test_split( valid_test_sequence_ids, test_size=0.50, random_state=42)现在,从 sequence_df 里抽出各个子集对应的行:
sequence_splits = { "train": sequence_df[sequence_df["EntryID"].isin(train_sequence_ids)], "valid": sequence_df[sequence_df["EntryID"].isin(valid_sequence_ids)], "test": sequence_df[sequence_df["EntryID"].isin(test_sequence_ids)],}for split, df in sequence_splits.items(): print(f"{split} has {len(df)} entries.")Output:
train has 3574 entries.valid has 1191 entries.test has 1192 entries.这样,我们就得到了一组干净、互不重叠的训练集、验证集和测试集。后面整个建模、调参与评估流程,都会围绕这三部分数据展开。
将蛋白质序列转换为平均嵌入
现在,我们把每个数据子集中的蛋白质序列,转换成对应的平均嵌入,就像前面已经演示过的那样。由于这一步可能比较耗时,尤其是在使用更大模型的时候,所以值得认真考虑怎样把流程做得更高效。使用 GPU 可以显著提速,但还有一个很重要的技巧:嵌入只计算一次,写入磁盘,之后需要时再直接加载,避免重复工作。
为了让这个流程更方便,我们先定义一对辅助函数,分别用于保存和加载序列嵌入:
def store_sequence_embeddings( sequence_df: pd.DataFrame, store_prefix: str, tokenizer: PreTrainedTokenizer, model: PreTrainedModel, batch_size: int = 64, force: bool = False,) -> None: """提取每条蛋白质序列的平均嵌入并保存到磁盘。""" model_name = str(model.name_or_path).replace("/", "_") store_file = f"{store_prefix}_{model_name}.feather" if not os.path.exists(store_file) or force: device = get_device() # 按批遍历蛋白质数据框,依次提取嵌入。 n_batches = ceil(sequence_df.shape[0] / batch_size) batches: list[np.ndarray] = [] for i in range(n_batches): batch_seqs = list( sequence_df["Sequence"][i * batch_size : (i + 1) * batch_size] ) batches.extend(get_mean_embeddings(batch_seqs, tokenizer, model, device)) # 把嵌入的每一维都单独存成数据框中的一列。 embeddings = pd.DataFrame(np.vstack(batches)) embeddings.columns = [f"ME:{int(i)+1}" for i in range(embeddings.shape[1])] df = pd.concat([sequence_df.reset_index(drop=True), embeddings], axis=1) df.to_feather(store_file)def load_sequence_embeddings( store_file_prefix: str, model_checkpoint: str) -> pd.DataFrame: """从磁盘加载已经保存好的嵌入数据框。""" model_name = model_checkpoint.replace("/", "_") store_file = f"{store_file_prefix}_{model_name}.feather" return pd.read_feather(store_file)现在,我们使用一个更强但也更耗计算的 ESM2 版本,它输出 640 维嵌入。然后通过 store_sequence_embeddings 函数,为每个子集保存对应的嵌入:
model_checkpoint = "facebook/esm2_t30_150M_UR50D"tokenizer = AutoTokenizer.from_pretrained(model_checkpoint)model = EsmModel.from_pretrained(model_checkpoint)for split, df in sequence_splits.items(): store_sequence_embeddings( sequence_df=df, store_prefix=assets(f"proteins/datasets/protein_dataset_{split}"), tokenizer=tokenizer, model=model, )嵌入保存好之后,后面任何时候需要都可以直接加载回内存。下面先看一眼模型真正会拿来训练的训练集长什么样:
train_df = load_sequence_embeddings( assets("proteins/datasets/protein_dataset_train"), model_checkpoint=model_checkpoint,)print(train_df)Output:
EntryID Sequence Length GO:0000166 GO:0000287 ... \0 A0A0C4DG62 MAHVGSRKRSRSRSRS... 218 0 0 ... 1 A0A1B0GTB2 MVITSENDEDRGGQEK... 48 0 0 ... 2 A0AVI4 MDSPEVTFTLAYLVFA... 362 0 0 ... ... ... ... ... ... ... ... 3571 Q9Y6W5 MPLVTRNIEPRHLCRQ... 498 0 0 ... 3572 Q9Y6W6 MPPSPLDDRVVVALSR... 482 0 0 ... 3573 Q9Y6Y9 MLPFLFFSTLFSSIFT... 160 0 0 ... ME:636 ME:637 ME:638 ME:639 ME:640 0 0.062926 0.040286 0.030008 -0.033614 0.023891 1 0.129815 -0.044294 0.023842 -0.020635 0.125583 2 0.153848 -0.075747 0.024440 -0.123321 0.020945 ... ... ... ... ... ... 3571 -0.001535 -0.084161 -0.014317 -0.141801 -0.040719 3572 0.120192 -0.086032 -0.016481 -0.108710 -0.077937 3573 0.114847 -0.028570 0.084638 0.038610 0.087047 [3574 rows x 946 columns]你会看到一大串名为 ME:1 到 ME:640 的列。它们表示的是 ESM2 模型最后一层隐藏状态经过 mean pooling 后得到的结果,本质上就是每条蛋白质序列的一个固定长度数值摘要。这些嵌入包含了模型在预训练过程中学到的生化与结构信息,也将作为后续分类器的输入特征。
这个数据框会被送进一个名为 convert_to_tfds 的函数。我们定义它,是为了更方便地为每个子集构建可训练的数据集:
import tensorflow as tfdef convert_to_tfds( df: pd.DataFrame, embeddings_prefix: str = "ME:", target_prefix: str = "GO:", is_training: bool = False, shuffle_buffer: int = 50,) -> tf.data.Dataset: """把嵌入数据框转换成 TensorFlow 数据集。""" dataset = tf.data.Dataset.from_tensor_slices( { "embedding": df.filter(regex=f"^{embeddings_prefix}").to_numpy(), "target": df.filter(regex=f"^{target_prefix}").to_numpy(), } ) if is_training: dataset = dataset.shuffle(shuffle_buffer).repeat() return dataset现在,用 convert_to_tfds 函数把训练集数据框转换成兼容 TensorFlow 的数据集:
train_ds = convert_to_tfds(train_df, is_training=True)从这些数据集中取出一个 batch 也很直接:先做 batch,再转成 NumPy 迭代器,然后调用 next 取出一批数据:
batch_size = 32batch = next(train_ds.batch(batch_size).as_numpy_iterator())batch["embedding"].shape, batch["target"].shapeOutput:
((32, 640), (32, 303))这个形状告诉我们:每个输入都是一个 640 维嵌入向量(来自 ESM2),而每个目标则是一个 303 维二值向量,用来表示每种分子功能是否存在。
提示
由于训练集里用了 .repeat(),它会不断循环数据并持续产出 batch。这对训练来说很方便,因为我们本来就希望多轮遍历训练集。相比之下,验证集和测试集不会重复,因此它们的 batch 最终会被取完,这正符合评估时的需求,因为每个样本应该只被看一次。
为了让整个数据集构建流程更简洁,我们把它又包装成一个统一的辅助函数 build_dataset:
def build_dataset( store_file_prefix: str, model_checkpoint: str) -> dict[str, tf.data.Dataset]: """从已保存的嵌入构建 train/valid/test 三个 TensorFlow 数据集。""" dataset_splits = {} for split in ["train", "valid", "test"]: dataset_splits[split] = convert_to_tfds( df=load_sequence_embeddings( store_file_prefix=f"{store_file_prefix}_{split}", model_checkpoint=model_checkpoint, ), is_training=(split == "train"), ) return dataset_splits这个函数会为三个数据子集都从磁盘中加载平均嵌入,并构建出可直接用于训练的 tf.data.Dataset 对象:
dataset_splits = build_dataset( assets("proteins/datasets/protein_dataset"), model_checkpoint=model_checkpoint)到这里,数据就算是彻底预处理完成,可以正式进入模型训练阶段了。
训练模型
现在,我们要在这些平均蛋白质嵌入之上训练一个简单的 Flax 线性模型。前面已经提到,原始蛋白质序列长度各不相同,但我们已经把它们统一转换成了固定长度嵌入。当前目标,是预测每个蛋白质会执行 303 种可能分子功能中的哪些。这是一个多标签分类(multilabel classification)问题,因为一个蛋白质可以同时带有多个功能标签。
在这个设定下,我们训练一个轻量级 MLP(multilayer perceptron,多层感知机),也就是一组带非线性的全连接层。需要强调的是,我们并不会微调原始 ESM2 模型;它保持冻结状态,我们训练的模型只是建立在它输出的嵌入之上。
模型代码如下:
import flax.linen as nnfrom flax.training import train_stateclass Model(nn.Module): """用于蛋白质功能预测的简单 MLP。""" num_targets: int dim: int = 256 @nn.compact def __call__(self, x): """将 MLP 层应用到输入特征上。""" x = nn.Sequential( [ nn.Dense(self.dim * 2), jax.nn.gelu, nn.Dense(self.dim), jax.nn.gelu, nn.Dense(self.num_targets), ] )(x) return x def create_train_state(self, rng: jax.Array, dummy_input, tx) -> TrainState: """初始化模型参数,并返回训练状态对象。""" variables = self.init(rng, dummy_input) return TrainState.create( apply_fn=self.apply, params=variables["params"], tx=tx )关于这个轻量模型,有几点值得说明:
它使用 nn.Sequential来堆叠各层结构,对这种简单模型来说,写法清晰而直观。激活函数使用的是 GELU(Gaussian Error Linear Unit),它是一种比 ReLU 更平滑的非线性函数。最后一层是一个 nn.Dense,把隐藏表示投影到功能标签数量(num_targets)上。它返回的是logits,不是概率,因此后面我们会在损失函数里配合合适的激活函数(例如sigmoid)把它转换成预测概率。这个模型是建立在冻结的 ESM2嵌入之上的,也就是说它不会更新 transformer 的权重,而只学习如何把固定嵌入映射到功能标签。这种做法高效、相对可解释,而且训练时内存占用也更低。
你可能也注意到了,我们还在模型类里附加了一个方便函数 create_train_state,专门用来创建训练状态。它把模型初始化、参数注册以及优化器配置,都封装进一个 TrainState 对象里。这样做尤其方便,因为当模型本身、用于推断形状的 dummy input 和优化器配置都已经在手边时,我们就能一次性把训练状态构造好。
现在根据训练数据框里有多少个 GO term 列,来实例化一个输出维度正确的模型:
targets = list(train_df.columns[train_df.columns.str.contains("GO:")])mlp = Model(num_targets=len(targets))到这里,模型已经准备好了,可以拿预计算好的嵌入作为输入,来学习预测一个蛋白质参与哪些分子功能。
定义训练循环
模型和数据都准备好之后,我们就可以定义“单步训练”函数了。一次训练步主要包括:
模型前向传播 计算损失 计算梯度 用梯度更新模型参数
具体实现如下:
@jax.jitdef train_step(state, batch): """执行一步训练,并更新模型参数。""" def calculate_loss(params): """根据 logits 计算 sigmoid 交叉熵损失。""" logits = state.apply_fn({"params": params}, x=batch["embedding"]) loss = optax.sigmoid_binary_cross_entropy(logits, batch["target"]).mean() return loss grad_fn = jax.value_and_grad(calculate_loss, has_aux=False) loss, grads = grad_fn(state.params) state = state.apply_gradients(grads=grads) return state, loss这里有两个关键点:
我们使用 sigmoid激活和二元交叉熵损失,这非常适合多标签分类。这里用的是sigmoid,而不是softmax,因为我们想对每一种可能蛋白质功能都做独立的“是 / 否”判断。别忘了,一个蛋白质可以同时具备多个功能。@jax.jit会把训练步骤编译起来,从而提高执行效率。
接下来,我们用 sklearn 里的工具定义一些评估指标,帮助我们不只盯着损失值,还能更全面地判断模型表现:
from sklearn import metricsdef compute_metrics( targets: np.ndarray, probs: np.ndarray, thresh=0.5) -> dict[str, float]: """计算 accuracy、recall、precision、auPRC 和 auROC。""" if np.sum(targets) == 0: return { m: 0.0 for m in ["accuracy", "recall", "precision", "auprc", "auroc"] } return { "accuracy": metrics.accuracy_score(targets, probs >= thresh), "recall": metrics.recall_score(targets, probs >= thresh).item(), "precision": metrics.precision_score( targets, probs >= thresh, zero_division=0.0, ).item(), "auprc": metrics.average_precision_score(targets, probs).item(), "auroc": metrics.roc_auc_score(targets, probs).item(), }我们会跟踪下面这些评估指标:
Accuracy:在所有标签上的正确预测比例。在这种类别极不平衡的多标签问题里,accuracy 往往会误导人,因为大多数标签都是 0,一个永远预测“没有该功能”的模型看起来也可能非常“准确”。不过它直观易懂,所以暂时还是保留。 Recall:真实正例中被模型正确找出来的比例,也就是真阳性 / 所有真实阳性。recall 高,说明模型不会漏掉太多真实功能。 Precision:模型预测为正的结果里,有多少是真的,也就是真阳性 / 所有预测为正。precision 高,说明模型不太容易乱报。 auPRC(precision-recall 曲线下面积):概括不同阈值下 precision 和 recall 的权衡。在像这里这种高度不平衡问题中,尤其有用。 auROC(receiver operating characteristic 曲线下面积):衡量模型在各种阈值下区分正负样本的能力。它是很常见的判别能力指标,但在高度不平衡数据里有时也会误导,因为它对正负类给予了相同权重。
在多标签场景下,我们会针对每个蛋白质功能(也就是每个 target / label)都计算这些指标,然后再取平均,得到一个更整体的模型表现视图。
这些评估指标会在 eval_step 中被调用:
def eval_step(state, batch) -> dict[str, float]: """执行评估步骤,并返回各目标平均后的指标。""" logits = state.apply_fn({"params": state.params}, x=batch["embedding"]) loss = optax.sigmoid_binary_cross_entropy(logits, batch["target"]).mean() target_metrics = calculate_per_target_metrics(logits, batch["target"]) metrics = { "loss": loss.item(), **pd.DataFrame(target_metrics).mean(axis=0).to_dict(), } return metricsdef calculate_per_target_metrics(logits, targets): """为一个多标签 batch 中的每个目标计算指标。""" probs = jax.nn.sigmoid(logits) target_metrics = [] for target, prob in zip(targets, probs): target_metrics.append(compute_metrics(target, prob)) return target_metrics这里的评估是在 batch 中按“每个蛋白质”来计算指标。对于每一条蛋白质记录,我们会:
先对它的 303 个 logits 应用 sigmoid,得到各功能的预测概率。再对这些概率设定阈值(例如 0.5),得到二值预测结果。 最后把它和真实功能标签进行比较,计算 accuracy、precision、recall、auPRC和auROC等指标。
然后对 batch 中的每个蛋白都重复一遍,最后再在蛋白之间对指标求平均。这告诉我们:模型对“每个蛋白质的一组功能标签”的预测能力如何。它并不是按 GO term 报告表现的。如果我们想看每个功能本身的表现,例如模型对 GO:0003677 的预测能力,就需要改成按列计算指标。
接下来,前面的组件会被整合进一个 train 函数中,而这种基本训练结构也会在后续章节反复出现。训练循环的核心逻辑是:先初始化训练状态,然后不断从数据集中取 batch 做训练,并且每隔一段时间进行一次评估:
def train( state: TrainState, dataset_splits: dict[str, tf.data.Dataset], batch_size: int, num_steps: int = 300, eval_every: int = 30,): """使用分批 TF 数据集训练模型,并记录性能指标。""" # 创建容器,用于保存训练和评估过程中计算出的指标。 train_metrics, valid_metrics = [], [] # 创建 batched 数据集,供每一步训练取用 batch。 train_batches = ( dataset_splits["train"] .batch(batch_size, drop_remainder=True) .as_numpy_iterator() ) steps = tqdm(range(num_steps)) # 带进度条的训练步数迭代器。 for step in steps: steps.set_description(f"Step {step + 1}") # 取出一批训练数据,转成 JAX 数组后执行训练。 state, loss = train_step(state, next(train_batches)) train_metrics.append({"step": step, "loss": loss.item()}) if step % eval_every == 0: # 对所有评估 batch 计算指标。 eval_metrics = [] for eval_batch in ( dataset_splits["valid"].batch(batch_size=batch_size).as_numpy_iterator() ): eval_metrics.append(eval_step(state, eval_batch)) valid_metrics.append( {"step": step, **pd.DataFrame(eval_metrics).mean(axis=0).to_dict()} ) return state, {"train": train_metrics, "valid": valid_metrics}关于这个训练循环,还可以补充几点:
高效的 batch 采样:训练数据通过 .as_numpy_iterator()以流式方式送入,而数据集中的.repeat()保证它会无限循环。定期评估:每隔 eval_every步,模型都会在完整验证集上评估一次,用前面定义好的指标(如auPRC、auROC)监控进展。指标聚合:验证指标先按 batch 计算,再通过 pd.DataFrame(...).mean(axis=0)在所有 batch 上取平均,这样可以得到整个验证集更稳定的表现估计。
现在开始真正训练模型。不过在这之前,先用一个非常实用的小技巧:为了避免你每次重跑代码单元时都从头训练一遍,我们引入 @restorable 装饰器。这个轻量工具会检查指定路径下是否已经存在训练好的模型;如果存在,它就会:
跳过重新训练; 把模型恢复成一个有效的 TrainState;返回模型以及已经保存好的指标。
这会让你的工作流更快,也更容易复现,尤其适合迭代开发和调试。用法如下:
import optaxfrom dlfb.utils.restore import restorable# 用单个 batch 的 dummy 数据初始化训练状态。rng = jax.random.PRNGKey(42)rng, rng_init = jax.random.split(key=rng, num=2)state, metrics = restorable(train)( state=mlp.create_train_state( rng=rng_init, dummy_input=batch["embedding"], tx=optax.adam(0.001) ), dataset_splits=dataset_splits, batch_size=32, num_steps=300, eval_every=30, store_path=assets("proteins/models/mlp"),)这里还有几个值得注意的参数,比如优化器(这里是 optax.adam)以及总训练步数(num_steps)。假设我们有大约 2,100 个训练样本,batch size 是 32,那么模型大概要经过 66 步才能把整个训练集看完一遍。设置 num_steps=300,意味着每个训练样本会被模型重复看到好几次。
模型训练完成之后,我们就可以像图 2-13 那样,来检查它的训练动态和在验证集上的表现:
import matplotlib.pyplot as pltimport seaborn as snsfrom dlfb.utils.metric_plots import DEFAULT_SPLIT_COLORSfig, ax = plt.subplots(nrows=1, ncols=2, figsize=(9, 4))# 绘制训练损失曲线。learning_data = pd.concat( pd.DataFrame(metrics[split]).melt("step").assign(split=split) for split in ["train", "valid"])sns.lineplot( ax=ax[0], x="step", y="value", hue="split", data=learning_data[learning_data["variable"] == "loss"], palette=DEFAULT_SPLIT_COLORS,)ax[0].set_title("训练步数上的损失变化")# 绘制验证指标曲线。sns.lineplot( ax=ax[1], x="step", y="value", hue="variable", style="variable", data=learning_data[learning_data["variable"] != "loss"], palette="Set2",)plt.legend(loc="center left", bbox_to_anchor=(1, 0.5))ax[1].set_title("训练步数上的验证指标变化");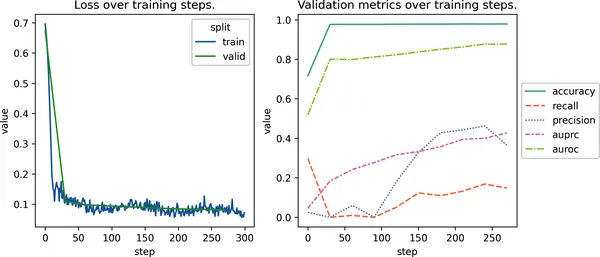
图 2-13. MLP 模型在 300 个训练步上的训练与评估结果。左图展示训练集和验证集的损失曲线,可以看到模型快速收敛,并在约 30 步后趋于稳定。右图展示了 auPRC、precision 和 recall 的逐步提升。由于类别极不平衡,accuracy 和 auROC 一直很高,但对这个问题的信息量其实不大。
在左图中,我们看到训练损失和验证损失都在最开始约 30 步内快速下降,随后趋于平稳。这是一条很典型的学习曲线,说明模型迅速收敛,而且没有出现明显不稳定现象(比如大幅震荡或发散)。这也说明:这个建立在冻结预训练嵌入之上的浅层 MLP,能够很快抓住数据中最容易学到的信号。
右图则跟踪了几个评估指标随时间的变化:
Accuracy和 auROC一开始就很高,而且变化不大,但在这种类别极不平衡的多标签问题里,它们其实容易误导。因为绝大多数功能标签都是负类(也就是某个蛋白并不具备绝大多数功能),所以一个主要预测 0 的模型也能拿到很高的这两个指标。因此,在这个场景里我们不会太依赖它们。auPRC一直在稳步提升,而且还没有完全平台化,这说明模型还在持续学会一些更细微的区分,理论上可能还能从更长训练中获益(也就是增加 num_steps)。precision 比 recall 提升得更快,这意味着模型对自己的预测越来越“谨慎而自信”,但仍然会漏掉一些真实正例。
综合来看,这些趋势说明:虽然大多数可学习信号在早期就已经被捕捉到,但在 recall 和 auPRC 上,模型可能仍然有进一步提升空间,尤其是在延长训练或换用更强架构的情况下。
提示
在每个训练循环里手工记录指标、再自己写可视化代码,多少有点繁琐。为了简化这个过程,后续章节会引入 MetricsLogger(负责记录数值)和 MetricsPlotter(负责绘图)。
除此之外,很多现代机器学习工作流都会使用托管式或自托管的 dashboard,自动收集、保存并实时展示指标。这些工具对监控实验、比较不同训练运行以及团队协作都很有帮助。常见选择包括:
TensorBoard Weights & Biases(W&B) MLflow
看到模型顺利训练成功、损失和指标曲线也朝着正确方向发展,当然很让人开心。但这还只是开始。真正有价值的洞察,来自于分析模型到底预测了什么、它在哪些地方做得好,以及它的局限在哪里。
查看模型预测
有了训练好的模型之后,接下来就是分析它的长处和短板。我们先为整个验证集生成预测结果,并把它们保存进一个数据框,方便后续检查:
valid_df = load_sequence_embeddings( store_file_prefix=f"{assets('proteins/datasets/protein_dataset')}_valid", model_checkpoint=model_checkpoint,)# 使用 batch size=1,避免最后余数样本被丢掉。valid_probs = []for valid_batch in dataset_splits["valid"].batch(1).as_numpy_iterator(): logits = state.apply_fn({"params": state.params}, x=valid_batch["embedding"]) valid_probs.extend(jax.nn.sigmoid(logits))valid_true_df = valid_df[["EntryID"] + targets].set_index("EntryID")valid_prob_df = pd.DataFrame( np.stack(valid_probs), columns=targets, index=valid_true_df.index)为了先从整体上感受模型表现,我们可以把完整预测矩阵画成热图。图 2-14 中,我们并排画出两张热图:左边是真实的蛋白质功能注释,右边是模型输出的预测概率。每一列对应一个蛋白质功能,每一行对应一个蛋白质:
fig, ax = plt.subplots(nrows=1, ncols=2, figsize=(11, 4))sns.heatmap( ax=ax[0], data=valid_true_df, yticklabels=False, xticklabels=False, cmap="flare",)ax[0].set_title("按蛋白质展示的真实功能注释")ax[0].set_xlabel("功能类别")sns.heatmap( ax=ax[1], data=valid_prob_df, yticklabels=False, xticklabels=False, cmap="flare",)ax[1].set_title("按蛋白质展示的预测功能注释")ax[1].set_xlabel("功能类别");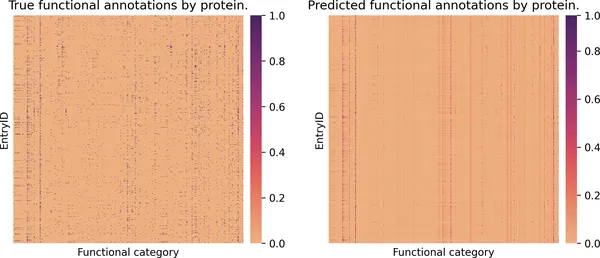
图 2-14. 蛋白质功能预测的热图总览。左图展示验证集中每个蛋白质的真实功能注释,右图展示模型给出的预测概率。两张矩阵都非常稀疏,而垂直条纹则对应那些较常见的功能标签。
这个可视化非常宏观,也比较“缩小视图”,但它能帮助我们建立对整体模型行为的直觉:
有些蛋白质功能在数据集中非常常见(会表现为明显的竖条纹),而模型通常也更容易把这些功能预测对。 稀有功能更难捕捉,模型经常会完全漏掉它们,因此预测热图里对应列可能很稀疏,甚至几乎空白。 还有少数功能会被过度预测,在很多蛋白上都出现淡淡的纵向色带,这说明模型对这些类别可能有点过度自信。 预测矩阵中很多格子呈现中间色调,这代表模型给出的概率并不极端,而是带有明显不确定性。
接下来,我们从这种定性观察转向定量分析,分别评估模型在每一种蛋白质功能上的表现:
metrics_by_function = {}for function in targets: metrics_by_function[function] = compute_metrics( valid_true_df[function].values, valid_prob_df[function].values )overview_valid = ( pd.DataFrame(metrics_by_function) .T.merge(go_term_descriptions, left_index=True, right_on="term") .set_index("term") .sort_values("auprc", ascending=False))print(overview_valid)Output:
accuracy recall precision auprc auroc \term GO:0004930 0.958858 0.000000 0.000000 0.948591 0.982272 GO:0004888 0.945424 0.177215 1.000000 0.849885 0.968354 GO:0003824 0.848027 0.731591 0.819149 0.849362 0.909372 ... ... ... ... ... ... GO:0003774 0.000000 0.000000 0.000000 0.000000 0.000000 GO:0051015 0.000000 0.000000 0.000000 0.000000 0.000000 GO:1902936 0.000000 0.000000 0.000000 0.000000 0.000000 description term GO:0004930 G protein-couple... GO:0004888 transmembrane si... GO:0003824 catalytic activity ... ... GO:0003774 cytoskeletal mot... GO:0051015 actin filament b... GO:1902936 phosphatidylinos... [303 rows x 6 columns]这个分析表明:模型在不同蛋白质功能上的表现差异非常大。例如,它在 GO:0004930(G protein-coupled receptor activity)这种功能上表现不错,但在 GO:0003774(cytoskeletal motor activity)等其他功能上就很吃力。不过,在解读这些结果时要保持谨慎:有些指标可能只建立在极少量验证样本之上,而那些在训练中本来就样本稀少的功能,表现差一些也完全在预期之中。某个高频功能得到高分,可能只是因为训练样本足够多;而稀有功能得分低,也未必令人意外。
阈值型指标与连续型指标
我们这里使用的评估指标,大体可以分成两类:阈值型(thresholded)和连续型(continuous)。 Precision 和 recall 是基于二值预测算出来的,也就是先对模型输出概率施加一个固定阈值(通常是 >0.5 )之后再计算。 auPRC (precision-recall 曲线下面积)和 auROC (receiver operating characteristic 曲线下面积)则与阈值无关。它们衡量的是:在所有可能阈值下,模型是否能把正样本排在负样本前面。 虽然听起来有点反直觉,但 precision 和 recall 完全可能为 0,而 auPRC 和 auROC 却仍然很高。这发生在这样一种情况下:模型确实把正确标签分到了更高概率,但这些概率始终没能超过判定阈值。于是,基于阈值的指标看起来像“完全失败”,而基于排序的指标仍然能反映出有意义的信号。 如果我们想改善当前这些阈值型指标,可以调低决策阈值,例如改成 0.2 或 0.3 ,从而鼓励模型做出更多正类预测。这个阈值也可以通过诸如 F1 score (precision 与 recall 的调和平均数)之类的指标自动调优。
接着,我们更仔细地看看:某个蛋白质功能在训练数据中出现得越频繁,模型在验证集上是不是也就越容易把它学会:
# 统计每种功能在训练集里出现的次数。overview_valid = overview_valid.merge( pd.DataFrame(train_df[targets].sum(), columns=["train_n"]), left_index=True, right_index=True,)print(overview_valid)Output:
accuracy recall precision auprc auroc \GO:0004930 0.958858 0.000000 0.000000 0.948591 0.982272 GO:0004888 0.945424 0.177215 1.000000 0.849885 0.968354 GO:0003824 0.848027 0.731591 0.819149 0.849362 0.909372 ... ... ... ... ... ... GO:0003774 0.000000 0.000000 0.000000 0.000000 0.000000 GO:0051015 0.000000 0.000000 0.000000 0.000000 0.000000 GO:1902936 0.000000 0.000000 0.000000 0.000000 0.000000 description train_n GO:0004930 G protein-couple... 138 GO:0004888 transmembrane si... 228 GO:0003824 catalytic activity 1210 ... ... ... GO:0003774 cytoskeletal mot... 5 GO:0051015 actin filament b... 17 GO:1902936 phosphatidylinos... 18 [303 rows x 7 columns]粗看之下,高表现功能(例如 auPRC 更高的功能)似乎确实也拥有更多训练样本。图 2-15 通过散点图更清楚地展示了这种关系:
fig = sns.scatterplot( x="train_n", y="auprc", data=overview_valid, alpha=0.5, s=50, color="grey")fig.set_xlabel("训练样本数")fig.set_ylabel("验证集 auPRC");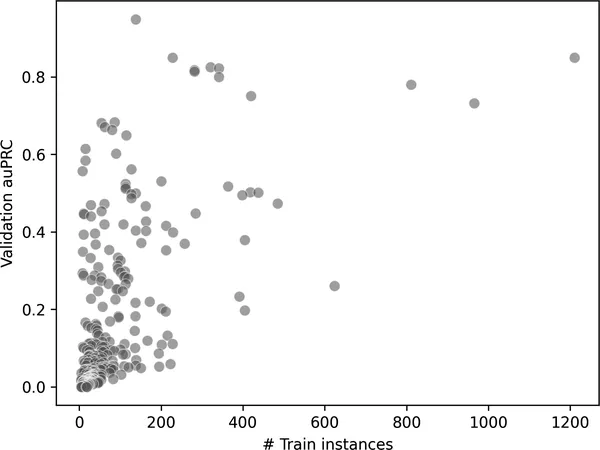
图 2-15. 不同蛋白质功能在训练集中出现频率,与其预测表现(auPRC)之间的关系。训练中更常见的功能,往往也会被模型预测得更准确。
这张图展示出一个很清楚的趋势:在训练集中出现越频繁的蛋白质功能,模型在验证集上通常也越容易预测准确(这里用 auPRC 衡量)。这完全符合我们的直觉,因为机器学习模型通常在样本充分的类别上表现更好。它也再次凸显了类别不平衡问题的挑战:稀有功能往往预测得很差,这未必是因为它们在生物学上更复杂,而可能只是因为模型根本没有足够数据去学习。
不过,另一个问题也随之而来:某个具体 auPRC 分数到底算不算“好”?例如,一个蛋白质功能的 auPRC = 0.8 听起来似乎不错,但它到底是不是优于随机?是否真的有意义?要回答这些问题,我们需要有一个可以对照的基线。
评估模型是否真的有用
为了让评估更有参照意义,我们把当前模型和两个非常简单的基线(baseline)做比较:
抛硬币基线(coin flip):对每个蛋白质功能都以相同概率随机预测 0或1。这代表“完全无知”的基准表现。按比例猜测(proportional guessing):对每个功能,以它在训练集里出现的频率作为预测为 1的概率。这代表模型只知道先验类别分布,但没有真正学到任何结构。
这些基线能帮助我们把模型表现放到具体语境中理解。如果训练出来的模型连这种简单启发式都赢不过,那就说明它很可能没有从数据中学到有意义的东西。
下面是两个基线的实现:
def make_coin_flip_predictions( valid_true_df: pd.DataFrame, targets: list[str]) -> pd.DataFrame: """为每个蛋白质功能生成随机抛硬币式预测。""" predictions = np.random.choice([0.0, 1.0], size=valid_true_df.shape) return pd.DataFrame(predictions, columns=targets, index=valid_true_df.index)def make_proportional_predictions( valid_true_df: pd.DataFrame, train_df: pd.DataFrame, targets: list[str]) -> pd.DataFrame: """按各功能出现频率生成随机预测。""" percent_1_train = dict(train_df[targets].mean()) proportional_preds = [] for target_column in targets: prob_1 = percent_1_train[target_column] prob_0 = 1 - prob_1 proportional_preds.append( np.random.choice([0.0, 1.0], size=len(valid_true_df), p=[prob_0, prob_1]) ) return pd.DataFrame( np.stack(proportional_preds).T, columns=targets, index=valid_true_df.index )这两个基线虽然简单,但能提供很有信息量的参考点。现在,我们把它们和训练好的模型放在一起:
prediction_methods = { "coin_flip_baseline": make_coin_flip_predictions(valid_true_df, targets), "proportional_guess_baseline": make_proportional_predictions( valid_true_df, train_df, targets ), "model": valid_prob_df,}接着,用和评估模型完全相同的方式来评估这些基线,也就是按蛋白计算指标,再取平均:
metrics_by_method = {}for method, preds_df in prediction_methods.items(): metrics_by_method[method] = pd.DataFrame( [ compute_metrics(valid_true_df.iloc[i], preds_df.iloc[i]) for i in range(len(valid_true_df)) ] ).mean()print(pd.DataFrame(metrics_by_method))Output:
coin_flip_baseline proportional_guess_baseline modelaccuracy 0.500916 0.956447 0.978569recall 0.499229 0.093555 0.128532precision 0.023883 0.079994 0.424301auprc 0.025307 0.039701 0.412350auroc 0.500027 0.535605 0.882679可以看到,我们的模型在所有指标上都明显优于这两个基线,尤其是在 precision、auPRC 和 auROC 上优势更加明显。这完全符合预期,因为训练模型真正利用了序列特征,而不只是盲猜。正如前面强调过的,在这个问题里 accuracy 并不是很可靠的指标;由于类别极度不平衡,哪怕是简单的按比例猜测,也能得到一个看起来颇高的 accuracy。
模型的大部分性能提升,主要来自 precision 的大幅提高,而 recall 的提升则相对有限。这意味着:当模型决定给出一个正类预测时,它通常比较准确;但与此同时,它也会漏掉不少真实正例。换句话说,它更偏向保守,而且倾向于预测“没有这个功能”。
这就体现出一个关键权衡:模型虽然保守,但比较准确。具体希望它表现成什么风格,要取决于你的应用需求。例如,如果你更在意不漏掉真实功能,就可以像前面提到的那样,通过降低决策阈值来提高 recall。
下一步,我们把模型的强项和弱项细化到“单个蛋白质功能”层面,并与两个基线逐项比较。这样就能看出:到底哪些功能预测得好,哪些功能预测得差。
auprc_by_function = {}for method, preds_df in prediction_methods.items(): metrics_by_function = {} for function in targets: metrics_by_function[function] = compute_metrics( valid_true_df[function], preds_df[function] ) auprc_by_function[method] = ( pd.DataFrame(metrics_by_function) .T.merge(go_term_descriptions, left_index=True, right_on="term") .set_index("term") .sort_values("auprc", ascending=False) )["auprc"].to_dict()图 2-16 用柱状图展示了“按功能划分的 auPRC 分数”,从而帮助我们看出模型最擅长处理哪些功能类别:
best_performing = ( pd.DataFrame(auprc_by_function) .merge(go_term_descriptions, left_index=True, right_on="term") .set_index("term") .sort_values("model", ascending=False) .head(20) .melt("description"))fig, ax = plt.subplots(figsize=(8, 5))sns.barplot( x="description", y="value", hue="variable", data=best_performing,)ax.set_title("模型表现最好的 20 个蛋白质功能")ax.set_ylabel("验证集 auPRC")plt.xticks(rotation=90);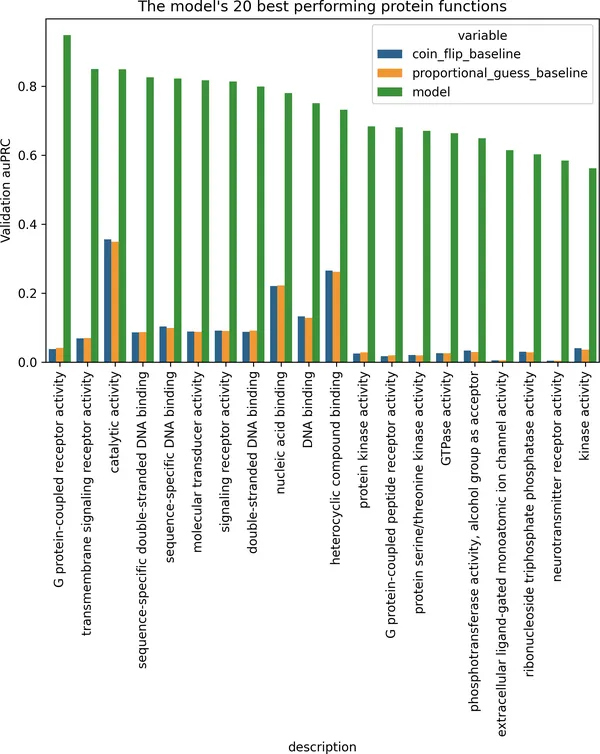
图 2-16. 按模型在验证集上的 auPRC 排序后,表现最好的 20 种蛋白质功能。图中的柱子同时展示了模型与两个简单基线(抛硬币、按比例猜测)之间的对比。
图中很多表现最好的功能,都和膜相关作用或信号传导相关,例如 GPCR activity、kinase activity、transmembrane receptor activity。一个可能的原因是:这些功能通常对应一些保守性较强的序列特征,例如跨膜螺旋区或催化结构域,因此模型更容易学到。虽然这仍带有推测性,但它和一个很合理的想法一致:凡是强烈依赖明确结构或生化模体(motif)的功能,往往会在序列层面呈现出更清晰的信号;而那些更依赖上下文的功能,则更难单靠序列本身判断。
综合这些结果来看,模型确实已经能够从某些类别的蛋白质功能中提取到有意义的生物学信号,而且它的表现明显优于简单基线。
在测试集上做最终检查
如果你想继续扩展和改进这个模型,可以先看看下一节的建议。等你对当前探索结果满意之后,就可以进入这个项目的最后一步:在测试集上做最终预测。记住,在项目的最后阶段之前,不要去碰测试集。
警告
一定要等到模型的所有内容都真正定稿之后,再去评估测试集,包括超参数、模型结构和训练策略都不再改动。反复在测试集上评估,会让结果变得过于乐观,从而损害结论的可信度。
下面我们用和验证集同样的方法,对测试集蛋白质做预测:
eval_metrics = []for split in ["valid", "test"]: split_metrics = [] for eval_batch in dataset_splits[split].batch(32).as_numpy_iterator(): split_metrics.append(eval_step(state, eval_batch)) eval_metrics.append( {"split": split, **pd.DataFrame(split_metrics).mean(axis=0).to_dict()} )print(pd.DataFrame(eval_metrics))Output:
split loss accuracy recall precision auprc auroc0 valid 0.080156 0.978457 0.126869 0.418515 0.411870 0.8808831 test 0.080675 0.978032 0.125820 0.435193 0.410439 0.879234测试集指标和验证集几乎一模一样,这是一个好现象。在很多工作流里,测试表现通常会比验证集略低一些,因为开发过程中会反复参考验证集,从而产生轻微过拟合。不过在这里,我们并没有做大量调优,所以两者差距非常小。由于测试集从头到尾都被严格留出,它给出的结果更能真实反映模型面对全新未见数据时的泛化能力。如果要对外报告结果,通常就会报告这组测试集指标。
改进与扩展
我们已经搭出了一个能工作的模型,它说明:只靠序列、结合预训练嵌入和轻量分类器,确实可以预测蛋白质功能。不过,这项工作还有很多可以继续改进、解释和扩展的方向。我们可以把这些想法大体分成两类:一类更偏分析与生物学洞察,另一类更偏机器学习方法上的改进。
但在真正投入技术升级之前,不妨先退一步,回到更大的问题上重新思考:
你为什么要做这件事? 最终谁会使用这个模型?他们真正需要的是什么? 现在这个原型能不能先拿给用户看,尽早收集反馈? 什么时候算“做完了”?当前模型是否已经够用? 哪些改进真的会显著提升它的实际价值? 这个任务或类似任务是否有已有 benchmark 可以参考? 你最在意的是什么?是否每一种功能都同样重要,还是你只关心某一类功能(例如酶与非酶)? 你是否需要可解释性?在某些应用里,理解“为什么模型会给出这个预测”,可能比把分数再提高一点更重要。
理想情况下,这些问题在建模开始前就应该考虑过;但即便现在再重新回顾一遍,它们依然能很好地帮助你决定下一步该往哪里走。
生物学与分析方向的探索
即便模型本身先保持不变,我们仍然可以通过探查它的行为、并将其与生物学预期对照,学到更多东西:
阈值调优(threshold tuning):我们的结果显示,在默认概率阈值 0.5下,模型的auPRC很高,但 recall 偏低。你可以尝试用F1 score之类的指标来优化阈值,无论是按功能单独调,还是全局统一调,从而找到 precision 和 recall 之间更好的平衡。跨物种泛化(species generalization):现在的数据集只包含人类蛋白,但这可能限制得太死了。可以尝试加入其他物种的蛋白-功能配对,看看模型表现是否会提升。 分析功能特异性的表现驱动因素:为什么模型在某些功能上表现很好(例如 GPCR activity),而在另一些功能上表现很差(例如 growth factor activity)?你可以去研究,功能出现频率、序列长度或其他属性,是否与预测表现相关。 考察蛋白质多功能性:模型是否更难处理“同时拥有很多功能”的蛋白质?可以按注释功能数对蛋白分组,再画出表现指标(例如 auPRC),看看是否存在趋势。挖掘可能“其实是真的”的假阳性:找出那些模型非常自信地预测出了某个功能、但标签里却没有该功能的蛋白质。有没有可能是模型说对了,只是数据库注释缺失了?如果要继续追踪验证,下一步该怎么做?
机器学习方向的改进
从机器学习角度出发,下面这些方向都值得进一步尝试:
调整 MLP 结构:当前模型只是建立在冻结嵌入之上的一个小型 MLP。你可以增加层数、加入 dropout,或者使用 batch normalization,在提升表达能力的同时控制过拟合。 替代性的输入编码:我们现在使用的是 mean-pooled embedding,这会丢失序列顺序信息。可以尝试在 token 级嵌入之上叠加 attention pooling,或者加一个小型 1D CNN / transformer。 特征工程:你可以在输入中额外加入蛋白长度、物种信息(如果你扩展到人类之外),甚至一些简单统计量,例如 embedding 范数。这些附加特征也许能帮助模型更有效地区分不同蛋白类型。 为每个功能单独训练 head:与其联合预测所有功能,不如为每个功能单独训练模型(或 head)。在任务高度不平衡或各任务关系不强时,这可能更有帮助。另一种思路是把 GO 功能先聚成若干簇,再为每一簇单独训练模型。 分层式功能预测:不要把每个功能都当作彼此独立的标签,而是利用 GO 层级结构,让预测过程也带层次感,比如先预测粗粒度功能,再细化到更具体功能。 尝试其他基础模型:你完全可以换用 Hugging Face 上的其他蛋白质语言模型,或者把多个模型的 embedding 拼接起来使用。 解冻语言模型: ESM2的嵌入是在通用任务上预训练出来的。如果直接对语言模型本体做蛋白质功能分类的微调,表现可能还会进一步提升,只是这样需要更多算力,也需要更复杂的训练设置。
注记
虽然不断往更复杂的模型上堆叠、追求更高分数很有诱惑力,但始终要让你的投入和项目的真实目标保持一致。很多时候,提升可解释性,或者扩展生物学覆盖面,可能比排行榜上再多涨一分更有价值。
小结
在这一章里,我们真正迈出了“用深度学习做生物学”的第一步。我们从一个人类蛋白质数据集出发,探索了如何利用预训练蛋白质语言模型提取有意义的表示,在这些表示之上训练一个简单分类器来预测蛋白质功能,并用定量指标评估模型表现。
在这个过程中,我们也碰到了生物学建模里非常典型的实际挑战:需要适应新的建模流程,要面对高度不平衡的标签分布,还必须非常谨慎地解读评估指标。
下一章里,我们会在这些基础之上继续推进,把关注点从蛋白质转向 DNA。你会从零开始在 Flax 中定义卷积神经网络,并端到端地训练它们,用来建模调控序列、预测功能元件,以及直接从基因组数据中发现 motif 模式。
━━ END ━━

