优秀学员R学习笔记(六):转录组数据的读取和处理
- 2026-05-29 19:09:56
R语言实战:转录组数据的读取和处理
0. 写在最前
在前面几篇笔记中我们更多着眼于基本概念和基础方法,而这篇笔记会开始将方向转变为在实战中可能遇到的场景,比如最基础的基因表达的数据该怎么导入、清洗以及针对我们的需求作出转换。
说到基因表达数据,我们一般就会想到Gene Expression Omnibus (GEO)数据库,官方网站:https://www.ncbi.nlm.nih.gov/geo/
点击进去后可以去搜索你感兴趣的疾病或者表型之类的,里面的信息包括数据集编号、标题、物种、样本数、类型等信息: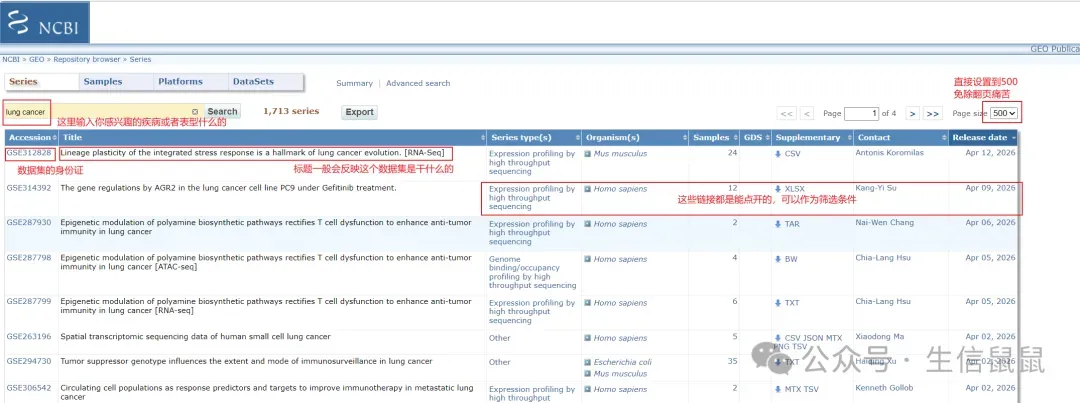
关于数据的种类需要说明:表达数据主要有这两种:高通量基因表达测序和基因表达芯片,前者又有bulk-RNA Seq(bulk的意思是大批的,也就是整个组织的)和sc-RNA Seq(也就是单细胞测序)之分
为了演示下载,我随便选了一个数据集,须注意图中对Series Matrix File的叙述: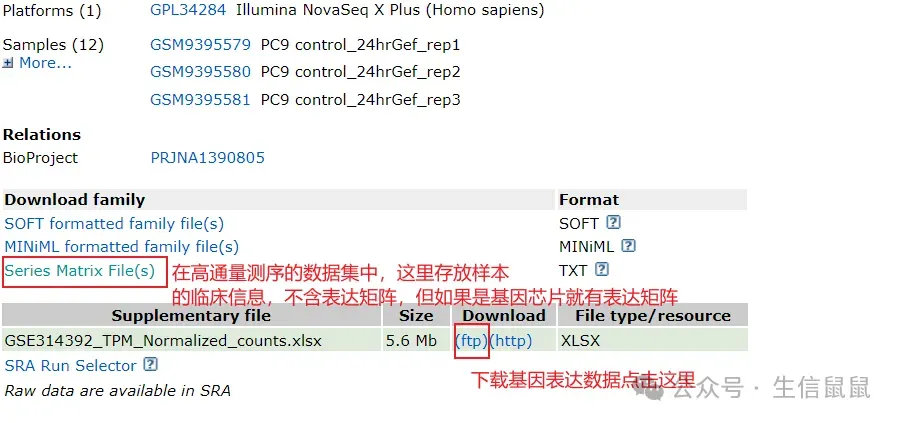
讲了这么多前面的废话,我们将以两个高通量测序数据集为案例来进行实战演练。 ——以下代码内容引用自生信技能树小洁老师
1. 实战演练一:相对乐观的数据情况
以数据集GSE211020为例,先下载下来原始文件尝尝咸淡: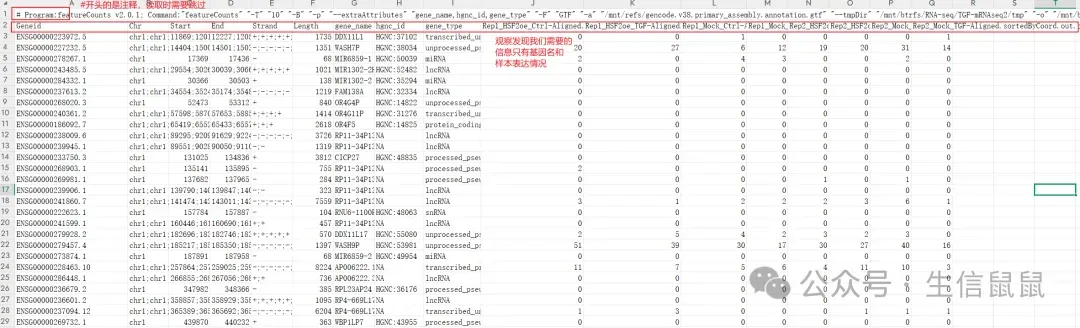
可以看到这个数据还是挺规整的,想要的基因名和样本名都在:我们只要取出这些构造表达矩阵就好
数据清洗部分
colnames(dat) # 看看列名(这一步可以帮助你定位到你想要的数据,这里我们看到了我们只要第7列(基因名)和10到17列(样本表达量)## [1] "Geneid" ## [2] "Chr" ## [3] "Start" ## [4] "End" ## [5] "Strand" ## [6] "Length" ## [7] "gene_name" ## [8] "hgnc_id" ## [9] "gene_type" ## [10] "Rep1_HSF2oe_Ctrl-Aligned.sortedByCoord.out.bam"## [11] "Rep1_HSF2oe_TGF-Aligned.sortedByCoord.out.bam" ## [12] "Rep1_Mock_Ctrl-Aligned.sortedByCoord.out.bam" ## [13] "Rep1_Mock_TGF-Aligned.sortedByCoord.out.bam" ## [14] "Rep2_HSF2oe_Ctrl-Aligned.sortedByCoord.out.bam"## [15] "Rep2_HSF2oe_TGF-Aligned.sortedByCoord.out.bam" ## [16] "Rep2_Mock_Ctrl-Aligned.sortedByCoord.out.bam" ## [17] "Rep2_Mock_TGF-Aligned.sortedByCoord.out.bam" exp <- dat[,c(7,10:17)] # 提取出我们想要的表达矩阵 library(stringr) colnames(exp) <- str_remove(colnames(exp),"-Aligned.sortedByCoord.out.bam") # Tidyverse内容,去掉没用的后缀# 也可以用str_split_i(colnames(exp),"-",1) library(dplyr) exp <- distinct(exp,gene_name,.keep_all = T) # 去除重复的基因名 exp <- tibble::column_to_rownames(exp,"gene_name") # 依旧是Tidyverse语法,把基因名这列作行名# 统计 exp 数据框中每一列的缺失值 (NA) 数量 colSums(is.na(exp)) # 结果全是0,就是没缺失## Rep1_HSF2oe_Ctrl Rep1_HSF2oe_TGF Rep1_Mock_Ctrl Rep1_Mock_TGF ## 0 0 0 0 ## Rep2_HSF2oe_Ctrl Rep2_HSF2oe_TGF Rep2_Mock_Ctrl Rep2_Mock_TGF ## 0 0 0 0为了匹配绘图或者其他分析对数据结构的要求,我们需要对数据进行转换 比如为了适配热图的输入要求,我们可以把数据转换为矩阵格式。
exp <- as.matrix(exp) # 很容易懂的函数 class(exp) # 结果是 matrix array## [1] "matrix" "array"# 我们要先对数据进行标准化,这里用edgeR包# 这里我们用CPM# CPM(Counts per million)仅对测序深度进行标准化,它将每个基因或转录本的表达水平标准化为每百万次测量次数的表达水平。主要用于比较不同样本之间的基因表达。 exp <- log2(edgeR::cpm(exp)+1)我们很多时候需要筛选满足某些条件的基因,比如我们筛选方差最大的3500个基因
对于这种大批量的数据,我们可以使用apply函数快速地进行处理(第四篇笔记中有过描述)
x <- sort(apply(exp,1,var)) # 计算所有基因的方差并排序,得到的是从小到大的结果 gs <- names(tail(x,3500)) # 那我们就从尾巴开始数出3500个基因# 这里的names函数是用来提取基因的名字的,因为apply函数生成的数据结构比较特殊 head(x,3) # 可以看到有文字和数值## MIR1302-2HG MIR1302-2 FAM138A ## 0 0 0 class(x) # 但x又是数值型,因此要用names()函数把这3500个基因的名字拿出来## [1] "numeric" exp_w <- exp[gs,] # 筛选符合条件的基因,构成新的表达矩阵在实际操作中,我们研究的通常是某种表型对某种疾病的影响,因此要筛选样本的分组以及和表型相关的基因。 以TGF处理vs对照 & 内质网应激为例
colnames(exp)## [1] "Rep1_HSF2oe_Ctrl" "Rep1_HSF2oe_TGF" "Rep1_Mock_Ctrl" "Rep1_Mock_TGF" ## [5] "Rep2_HSF2oe_Ctrl" "Rep2_HSF2oe_TGF" "Rep2_Mock_Ctrl" "Rep2_Mock_TGF"# 这里去除重复组的标识 Group <- colnames(exp) %>% str_remove("Rep1_|Rep2_")#通配符:\\d代表所有数字(1位),\\d+代表1位或多位数字# colnames(exp) %>% # str_remove("Rep\\d+_") %>% # 统计分组的数量 table(Group)## Group## HSF2oe_Ctrl HSF2oe_TGF Mock_Ctrl Mock_TGF ## 2 2 2 2---# %in% 划定范围,按照列取子集 k1 <- Group %in% c("HSF2oe_Ctrl","HSF2oe_TGF");table(k1)## k1## FALSE TRUE ## 4 4# 关键词匹配,HSF,和k1是等价的 k2 <- str_detect(Group,"HSF2");table(k2)## k2## FALSE TRUE ## 4 4 exp_h <- exp[,k1] ergenes <- data.table::fread("ergenes.txt",data.table = F, header = F # 这里防止把第一行当作列名 )# 读取内质网应激相关基因 head(ergenes) #可以看到V1列就是基因名单## V1## 1 HSPA5## 2 EIF2AK3## 3 XBP1## 4 ERN1## 5 ATP2A2## 6 ATF6#exp_h[ergenes$V1,] # 直接这么做会报错,因为有一些相关基因不存在于表达矩阵中 k3 = rownames(exp_h) %in% ergenes$V1;table(k3) # 写个条件判断,筛选出矩阵中存在的相关基因## k3## FALSE TRUE ## 58645 783 exp_h = exp_h[k3,]那假如我之后要对这些基因用ggplot2绘图,我就得把它转化成长数据(现在很明显是宽数据)
df_h = as.data.frame(exp_h)# 变回数据框 df_h = tibble::rownames_to_column(df_h,"geneid") # 把基因名这列加回来 library(tidyr) test1 <- pivot_longer(data = df_h, cols = -geneid, # 排除geneid列 names_to = "sample_nm", # 其他列全部作为新的sample_nm列的内容 values_to = "exp") # 原来的表达值单独在exp列展开 head(test1) ## # A tibble: 6 × 3## geneid sample_nm exp## <chr> <chr> <dbl>## 1 UBE2J2 Rep1_HSF2oe_Ctrl 6.52## 2 UBE2J2 Rep1_HSF2oe_TGF 6.53## 3 UBE2J2 Rep2_HSF2oe_Ctrl 6.54## 4 UBE2J2 Rep2_HSF2oe_TGF 6.40## 5 RER1 Rep1_HSF2oe_Ctrl 7.34## 6 RER1 Rep1_HSF2oe_TGF 7.28然后我们就可以选择我们感兴趣的基因做可视化
genes = c("JUN","RER1") # 定义你想要做可视化的基因 k4 = test1$geneid %in% genes;table(k4) # 条件筛选## k4## FALSE TRUE ## 3124 8 pdat = test1[k4,] pdat = mutate(pdat, Group = str_split_i(pdat$sample_nm,"_",3)) # 因为我只想知道它是TGF还是Control library(ggplot2) library(paletteer) ggplot(pdat,aes(x = geneid, y = exp, fill = Group))+ geom_boxplot()+ scale_fill_paletteer_d("ggsci::category20c_d3")+ theme_bw()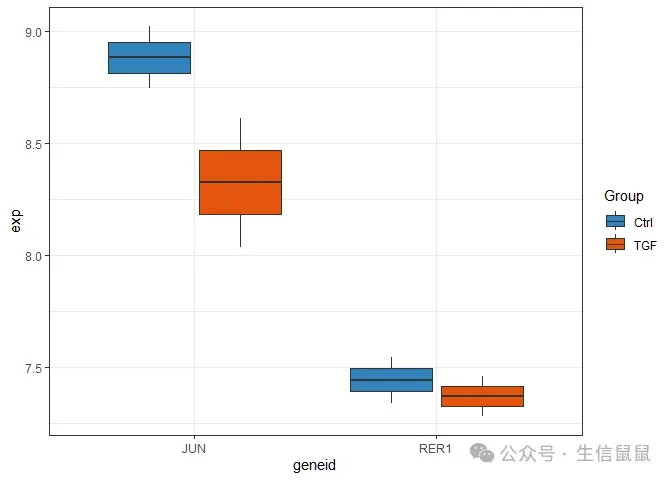
pdat2 = as.data.frame(t(exp_h[genes,])) pdat2 = mutate(pdat2, Group = str_split_i(rownames(pdat2),"_",3)) library(ggplot2) library(paletteer) library(ggExtra) p1 <- ggplot(pdat, aes(x = geneid, y = exp, fill = Group)) +# 2. 绘制半透明箱线图,并隐藏自带的离群点(防止与后面的散点重叠画两次) geom_boxplot(alpha = 0.6, outlier.shape = NA, position = position_dodge(width = 0.8)) +# 3. 叠加抖动散点,映射边框颜色,使其与箱线图完美对齐 geom_point(aes(color = Group), position = position_jitterdodge(jitter.width = 0.2, dodge.width = 0.8), size = 1.5, alpha = 0.8) +# 4. 统一填充色和边框色的调色板 scale_fill_paletteer_d("ggsci::category20c_d3") + scale_color_paletteer_d("ggsci::category20c_d3") +# 5. 完善标题和轴标签 labs(x = "Gene", y = "Expression Level", title = "Expression of JUN and RER1") +# 6. 使用最干净的经典主题(去网格线,保留 L 型坐标轴) theme_classic(base_size = 14) +# 7. 细节打磨 theme( plot.title = element_text(hjust = 0.5, face = "bold"), # 标题居中加粗 legend.position = "top", # 图例放顶部更省空间 legend.title = element_blank(), # 隐藏图例小标题 "Group" axis.text.x = element_text(face = "italic", color = "black"), # 基因名按规范斜体 axis.text.y = element_text(color = "black") )print(p1)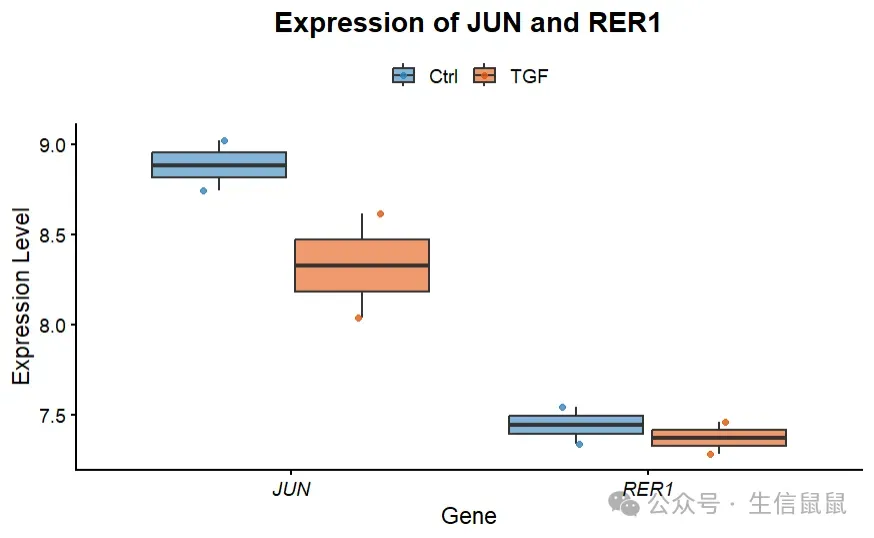
# 接下来因为要画散点图,对数据进行转置(行列交换) pdat2 = as.data.frame(t(exp_h[genes,])) head(pdat2)## JUN RER1## Rep1_HSF2oe_Ctrl 9.021863 7.340520## Rep1_HSF2oe_TGF 8.614397 7.283544## Rep2_HSF2oe_Ctrl 8.744060 7.545110## Rep2_HSF2oe_TGF 8.037100 7.458535 pdat2 = mutate(pdat2, Group = str_split_i(rownames(pdat2),"_",3)) # 还是只在意处理方式# 1. 绘制带有回归线的主体散点图 p2 <- ggplot(pdat2, aes(x = JUN, y = RER1, fill = Group, color = Group)) +# 2. 添加线性回归趋势线 (lm),带半透明的置信区间 geom_smooth(method = "lm", se = TRUE, alpha = 0.15, show.legend = FALSE) +# 3. 使用带黑色描边的实心点 (shape = 21),这是提升质感的绝对利器 geom_point(shape = 21, size = 3.5, color = "white", stroke = 0.5, alpha = 0.85) + scale_fill_paletteer_d("ggsci::category20c_d3") + scale_color_paletteer_d("ggsci::category20c_d3") + labs(x = "JUN Expression", y = "RER1 Expression", title = "Correlation between JUN and RER1") + theme_classic(base_size = 14) + theme( plot.title = element_text(hjust = 0.5, face = "bold"), legend.position = "bottom", legend.title = element_blank(), axis.text = element_text(color = "black") )# 4. 召唤终极武器:将主体图传入 ggMarginal 添加边缘密度分布图 p2_final <- ggMarginal( p2, type = "density", # 选择密度图 (也可选 histogram) groupColour = TRUE, # 边缘图轮廓按组着色 groupFill = TRUE, # 边缘图内部按组填充 alpha = 0.4 # 边缘图透明度 ) p2_final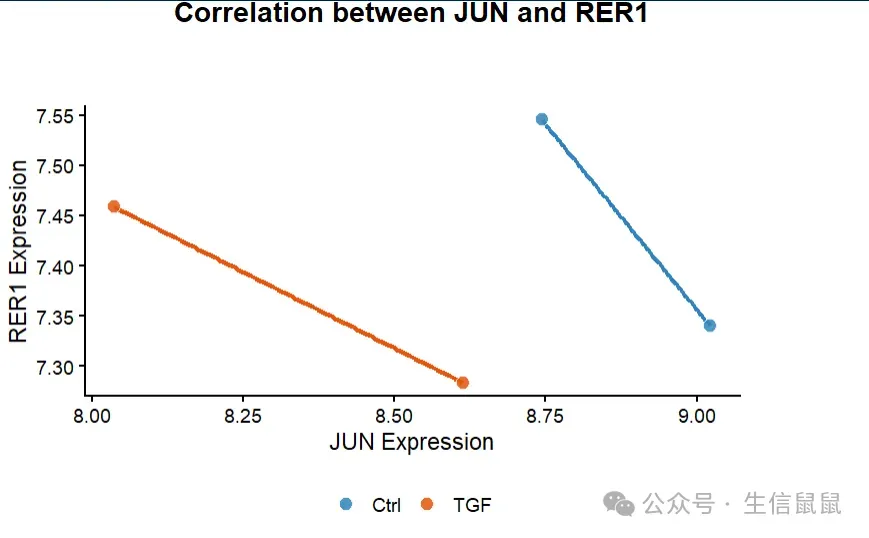
2. 实战演练二:比较阴间的数据情况
能够像上一个数据集那样做完分析那当然是皆大欢喜,但如果有一些比较坑的数据集该怎么办?比如GSE126848数据集,我们先打开看看: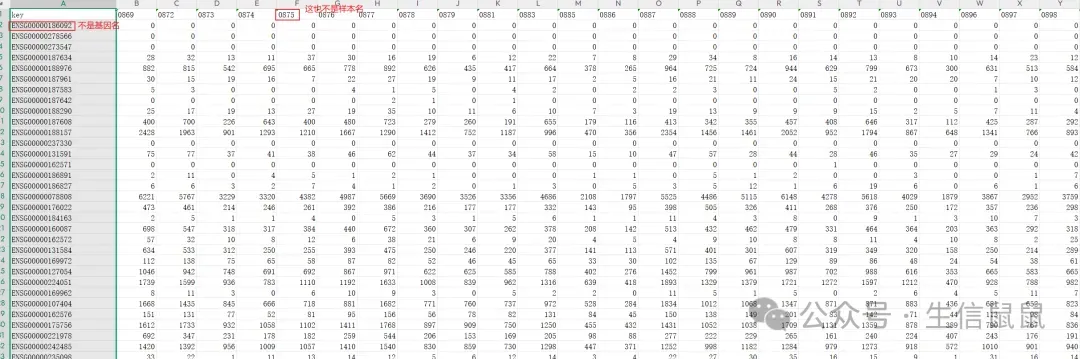
我说白了这个真的拉完了,基因名和样本名都没有,到底想咋滴?
但是我们先操作一下试试看呢? 依旧先读取数据
rm(list=ls()) dat = data.table::fread("GSE126848_Gene_counts_raw.txt.gz", data.table = F,header = T) dat = tibble::column_to_rownames(dat,"key")依旧清洗
colnames(dat)## [1] "0869" "0872" "0873" "0874" "0875" "0876" "0877" "0878" "0879" "0881"## [11] "0883" "0885" "0886" "0887" "0888" "0889" "0890" "0891" "0892" "0893"## [21] "0894" "0896" "0897" "0898" "0899" "0910" "2683" "2684" "2685" "2687"## [31] "2688" "2689" "2691" "2692" "2693" "2696" "2697" "2698" "2701" "2703"## [41] "2704" "2705" "3992" "3993" "3994" "3995" "3996" "3997" "3998" "4000"## [51] "4002" "4004" "4005" "4006" "4007" "4008" "4010" exp = dat# 统计 exp 数据框中每一列的缺失值 (NA) 数量 colSums(is.na(exp))## 0869 0872 0873 0874 0875 0876 0877 0878 0879 0881 0883 0885 0886 0887 0888 0889 ## 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 ## 0890 0891 0892 0893 0894 0896 0897 0898 0899 0910 2683 2684 2685 2687 2688 2689 ## 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 ## 2691 2692 2693 2696 2697 2698 2701 2703 2704 2705 3992 3993 3994 3995 3996 3997 ## 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 ## 3998 4000 4002 4004 4005 4006 4007 4008 4010 ## 0 0 0 0 0 0 0 0 0还是先标准化一下:
exp = as.matrix(exp) library(tinyarray)# 小洁老师做的Tinyarray包,直接把ENSGxxxxxx转化成基因名 exp = trans_exp_new(exp) # 转化 class(exp)## [1] "matrix" "array" exp = log2(edgeR::cpm(exp)+1)基因筛选不再赘述,直接来到最恶心的样本信息提取环节
library(tinyarray) # 还是这个包可以帮我们提取数据集的临床信息 pd = geo_download("GSE126848")$pd head(pd,3)## title description## GSM3615293 NAFL_1 2683## GSM3615294 NAFL_2 2685## GSM3615295 NAFL_3 2687## relation## GSM3615293 BioSample: https://www.ncbi.nlm.nih.gov/biosample/SAMN10979953## GSM3615294 BioSample: https://www.ncbi.nlm.nih.gov/biosample/SAMN10979952## GSM3615295 BioSample: https://www.ncbi.nlm.nih.gov/biosample/SAMN10979951## relation.1## GSM3615293 SRA: https://www.ncbi.nlm.nih.gov/sra?term=SRX5401559## GSM3615294 SRA: https://www.ncbi.nlm.nih.gov/sra?term=SRX5401560## GSM3615295 SRA: https://www.ncbi.nlm.nih.gov/sra?term=SRX5401561## characteristics_ch1.2 characteristics_ch1## GSM3615293 disease: NAFLD gender: Male## GSM3615294 disease: NAFLD gender: Male## GSM3615295 disease: NAFLD gender: Male# 这里发现title才是我们想要的样本名字,之前奇怪的数字是description# 所以我们只是要title和description这两列可以得到一比一转换就可以# 然而pd和exp顺序必须要匹配 rownames(pd) = NULL # 先把临床信息的行名给挪走 pd = column_to_rownames(pd,"description") # 再把description拿出来当列名不是吧, 不会真的有人以为已经结束了吧 最恶心的细节在这:
再回顾我们刚才看到的表达矩阵
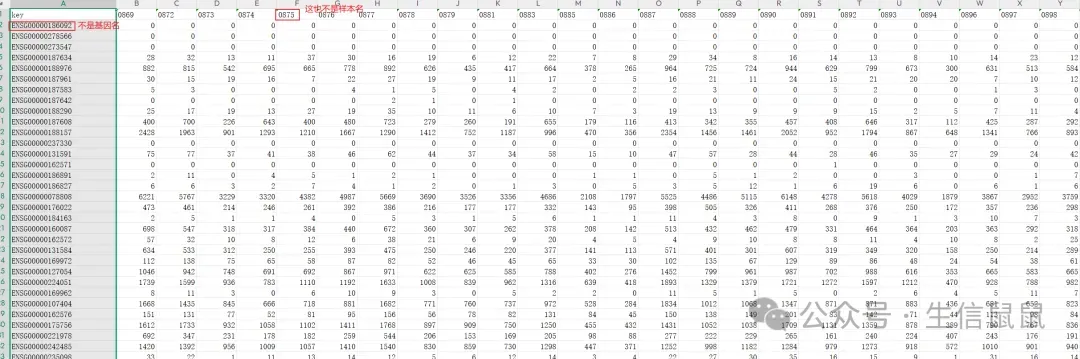
你会发现这些0开头的四位数,在临床信息里只有三位数 这个要用到条件判定来加上
k = str_length(rownames(pd))==3;table(k) # 条件筛选行名长度只有3的,也就是我们要转换的description## k## FALSE TRUE ## 31 26 rownames(pd) = ifelse(k, paste0("0",rownames(pd)), rownames(pd)) # 给符合条件的前面加个0#取交集函数intersect s = intersect(rownames(pd),colnames(exp)) pd = pd[s,] exp = exp[,s] rownames(pd) = pd$title colnames(exp) = pd$title colnames(exp)## [1] "NAFL_1" "NAFL_2" "NAFL_3" "NAFL_4" ## [5] "NAFL_5" "NAFL_6" "NAFL_7" "NAFL_8" ## [9] "NAFL_9" "NAFL_10" "NAFL_11" "NAFL_12" ## [13] "NAFL_13" "NAFL_14" "NAFL_15" "NASH_1" ## [17] "NASH_2" "NASH_3" "NASH_4" "NASH_5" ## [21] "NASH_6" "NASH_7" "NASH_8" "NASH_9" ## [25] "NASH_10" "NASH_11" "NASH_12" "NASH_13" ## [29] "NASH_14" "NASH_15" "NASH_16" "Normal-weight_1" ## [33] "Normal-weight_2" "Normal-weight_3" "Normal-weight_4" "Normal-weight_5" ## [37] "Normal-weight_6" "Normal-weight_7" "Normal-weight_8" "Normal-weight_9" ## [41] "Normal-weight_10" "Normal-weight_11" "Normal-weight_12" "Normal-weight_13"## [45] "Normal-weight_14" "Obese_1" "Obese_2" "Obese_3" ## [49] "Obese_4" "Obese_5" "Obese_6" "Obese_7" ## [53] "Obese_8" "Obese_9" "Obese_10" "Obese_11" ## [57] "Obese_12" Group = pd$title %>% # 我们把平行对照都删掉 str_remove("_\\d+") Group %>% # 统计分组的数量 table()## .## NAFL NASH Normal-weight Obese ## 15 16 14 12# %in% 划定范围,安列取子集 k1 = Group %in% c("NAFL","NASH");table(k1)## k1## FALSE TRUE ## 26 31 exp_h = exp[,k1] s = intersect(rownames(pd),colnames(exp_h)) pd = pd[s,] exp_h = exp_h[,s] ergenes = data.table::fread("ergenes.txt",data.table = F, header = F)#exp_h[ergenes$V1,] #有一些基因不存在与表达矩阵中 k3 = rownames(exp_h) %in% ergenes$V1;table(k3)## k3## FALSE TRUE ## 18929 781 exp_h = exp_h[k3,]后面的分析就一模一样了,这里不再赘述了。
3. 后记
其实写到这里再回忆之前踩过的坑就气不打一处来,但是这些问题总归是可以慢慢解决的。 还请大家多多交流、指正,希望能帮到各位读者,谢谢各位!
By 鼠鼠

