刚开始接触生物制品审评路径时,应该大概率都遇到过这些困惑:同样是生物制药,为什么有的是BLA,有的是NDA?为什么胰岛素的生物类似药在2020年前后走的是不同的申报通道?为什么同样是重组蛋白,有些归CDER管、有些归CBER管?
这一连串问题的答案都指向同一个原因:美国生物制药的监管不是建在一套法律上,而是建在两套法律上。 这两套法律:FD&C Act和PHS Act,有不同的历史起点、不同的逻辑起点,以及至今仍在演化的管辖边界。
本书的第二章标题为《Regulatory Pathways Impacting Biopharmaceuticals》,作者从监管路径的历史沿革讲起,先梳理美国体系,再到欧洲法规,最后对两者进行全方位比较。本期我们先聚焦美国法规部分的核心:FD&C Act和PHS Act这两套法源是怎么来的,它们各自管什么,NDA和BLA的两个路径差异是什么,以及为什么CMC从业者需要理解这个框架。
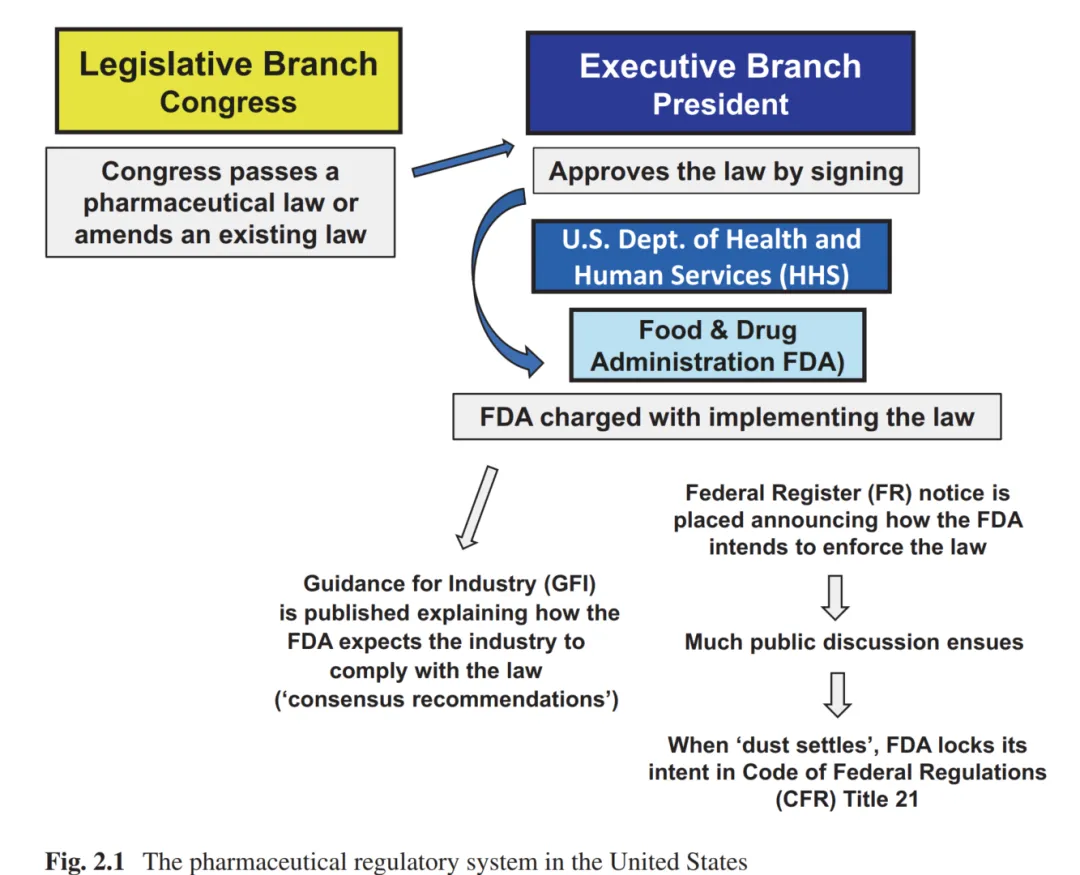
作者先用了一张简洁明了的逻辑图梳理了美国的制药监管体系,其主要路线关系为:国会立法→HHS(卫生与公众服务部)→FDA(食品和药品管理局)执行,三者各司其职。
图片来源:Geigert, The Challenge of CMC Regulatory Compliance for Biopharmaceuticals, 4th ed., Chap. 2
而在这一套运行规则中,有三个层级的实用文件需要注意,即CFR(Code of Federal Regulations),FR(Federal Register)和GFI(Guidance for Industry)。其中CFR为具有法律约束力的法规汇编,告诉你法规底线在哪;GFI为FDA发布的行业指导文件,告诉你最佳路径怎么走;FR为联邦公报,法规提案和公告的官方期刊,告诉你明天可能会改什么。 三个信息源的结合才能有效形成完整的CMC合规判断。
在理解了监管体系的层级后,再往下看两条法律的后续各自申报的路径。
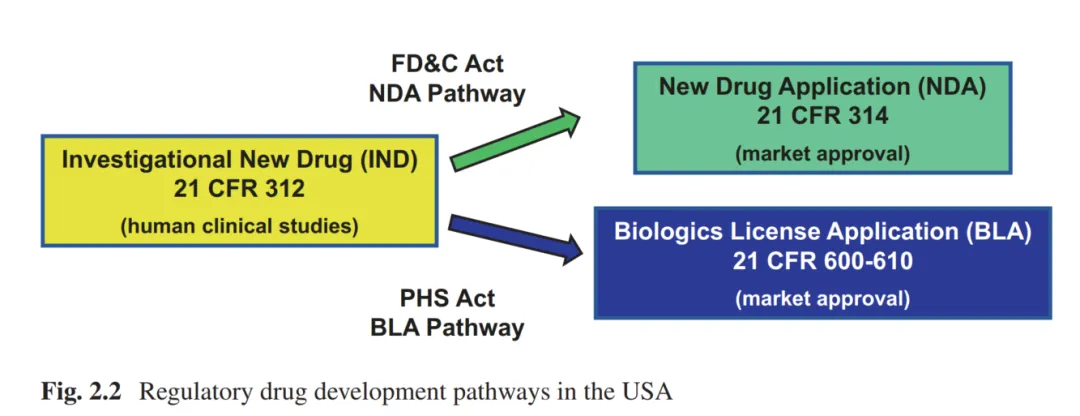
图片来源:Geigert, The Challenge of CMC Regulatory Compliance for Biopharmaceuticals, 4th ed., Chap. 2
从图示可以看出两条路径的起点都是IND(对应CFR为21 CFR 312),由FD&C Act管理的则为NDA(New Drug Application)路径,上市遵循21 CFR 314;而由PHS Act管理的则为BLA(Biologics License Application)路径,上市遵循21 CFR 600-610。
同时作者给出了一个2×2矩阵的法律矩阵,即两套法律,两个审评中心。FDA内部的两个审评中心分别为:CDER(药品审评与研究中心)和CBER(生物制品审评与研究中心)。而在后续的实际操作中,2003年CDER从CBER手中接管了治疗性蛋白和单抗的审评权,所以这个“法源-中心”的对应关系到今天已经不完全一一对应,这个演变的细节会在后续章节逐步展开。
作者在介绍FD&C Act时,先从法规结构讲起,然后排列一条清晰的历史时间线。
FD&C Act首创的路径是IND → NDA。IND相关要求编在21 CFR Part 312,NDA相关要求在21 CFR Part 314。作者在书中给出了两个法规在官网的部分条款目录,涵盖了IND的提交要求、临床分期、临床暂停等,以及NDA的申请内容格式、补充变更等,这些都是在后续CMC申报绕不开的关键点。
而真正让FD&C Act和生物制药产生关联的,是1941年的《胰岛素修正案》(Insulin Amendment Act)。作者对这段历史的描述是:
“In 1941, Congress passed the Insulin Amendment Act to ensure that the protein hormone, porcine insulin, could also be regulated by the FDA under the FD&C Act. This amendment to the law, allowed over time, the ‘grandfathering in’ of over 100 other proteins (both natural-sourced and recombinant), specifically proteins that were hormones or enzymes.”
这个“grandfathering in”是理解这段历史的关键词。所谓“祖父条款”(grandfather clause),在法律语境里指新法规生效后,允许某些已有产品或行为继续按旧规则运行,而不必满足新法规的全部要求。就FD&C Act而言,1938年这部法律本来是为化学药设计的——但1941年的胰岛素修正案开了一道门,让后来超过100种蛋白质(包括天然来源和后来出现的重组蛋白)也沿着NDA这条路走了几十年。
所以当翻看某些老产品的审评历史时,偶尔会遇到一个看似不合逻辑的情况:一个明明是重组蛋白的生物药,走的是NDA。而其原因并不是它被“被错误分类”,而是“被历史路径锁定”了。
这些”被错误分类“的生物制药,在2020年3月23日才终于被改写。作者原文为:
“It was not until March 23, 2020, that all of these proteins were transferred out of the FD&C law and moved over into the Public Health Services (PHS) law.”
即此前被“祖父条款”保护、沿NDA路径监管的所有蛋白质产品(包括重组人胰岛素、重组人生长激素、重组人透明质酸酶等)在这一天被正式移入PHS Act的管辖范围。
值得注意的是,作者在这一节末尾给出了一个关于蛋白质组成非常重要的边界定义:
“Proteins (greater than 40 aa) are considered biologicals regulated under the PHS Act. Peptides (no more than 40 amino acids) are considered chemical drugs regulated under the FD&C Act. However, if the peptide is to be used as a vaccine, it will be regulated under the PHS Act.”
即超过40个氨基酸组成的蛋白质将归于PHS Act下管理,而不超过40个氨基酸组成的多肽,则被视为化学药,归于FD&C Act下监管,除非其应用于疫苗领域。因此这个40个氨基酸的边界,直接决定了一个产品在FDA的申报路径归属。
1938年FD&C Act通过时,国会对化学药已经有了监管框架,但很快发现有一类在当时被称为‘biologicals’的药品用同一套规则管不住。它们和普通化学药不一样,它们的纯度通常不高,制造工艺需要更多监管,产品放行也需要额外的检测。于是短短六年后,即1944年,《公共健康服务法》(PHS Act)诞生了,并创立了专属路径:IND → BLA。
BLA相关要求编在21 CFR Part 600-610,作者在书中给出了官网的部分条款目录。而其中IND部分(21 CFR 312)和FD&C Act完全相同,即两条法律共享同一套临床研究管理规则。
关于BLA这个名字,作者补充了一个命名演变:最初PHS Act下的上市许可叫PLA/ELA(Product License Application / Establishment License Application),1996年才简化为现在的BLA。
真正体现这部法律底层逻辑的,是它对“biological product”的定义。作者把1944年的原版定义和当今的现行定义并列展示:
1944年版:
“a virus, therapeutic serum, toxin, antitoxin or analogous product, or arsphenamine or derivative of arsphenamine (or any other trivalent organic arsenic compound)…”
“a virus, therapeutic serum, toxin, antitoxin, vaccine, blood, blood component or derivative, allergenic product, protein or analogous product, or arsphenamine or derivative of arsphenamine…”
对比一下就能看出几个关键变化:加了疫苗(vaccine)、血液制品(blood, blood component or derivative)、过敏原产品(allergenic product),以及最关键的——protein。
而对于第四波的基因治疗产品(AAV载体、CAR-T等),作者给出的定位是:
“gene therapy-based biopharmaceuticals are currently embraced under the definition of ‘analogous products’”
即在作者成书时,基因治疗产品被归在“类似产品(analogous products)”这个筐里。作者在后续章节中会提到,FDA已经为这类产品设立了专门的审评办公室(OTP),说明分类体系还在持续细化中。
作者在这一节末尾专门用了一段来讲PHS Act定义中两个有趣的历史细节:
砷凡纳明(arsphenamine)至今还在定义里。
“Surprisingly, the chemically synthesized arsenic-containing organic compound, arsphenamine (also known as Compound 606), still remains today in the legal definition of a biological product in the PHS Act. Most likely it was included in the 1944 definition of a biological because it was the first chemically synthesized antibiotic, as well as the only effective treatment for syphilis at that time.”
砷凡纳明(Compound 606)是1910年代Paul Ehrlich开发的抗梅毒药物,也是世界上第一个化学合成的抗生素。1944年起草PHS Act时,它是当时为数不多的靶向性治疗药物之一,加之生产过程中需要生物活性检测来确保效力和安全性,因此被纳入了生物制品的法律定义。
80年过去了,这个19世纪的砷化合物依然躺在美国的生物制品定义里,既是一段药物史的化石,也是法规演变的活见证。
“In 2010, when ‘protein’ was added to the list of biological products in the PHS Act, it had the phrase ‘protein (except any chemically synthesized polypeptides)’; today, the phrase is only ‘protein’. This leaves open the possibility that if a protein could be commercially chemically synthesized it would be included under the PHS Act umbrella.”
2010年“protein”被正式写入PHS Act时,原文有一个括号补充为“except any chemically synthesized polypeptides”(化学合成的多肽除外)。但到了今天,这个括号被拿掉了,定义里只剩一个“protein”。
这个改动的含义是:若一个大于40个氨基酸的蛋白质可以通过化学合成实现商业化生产,它在法律上就可能被归为PHS Act管辖的生物制品,而不再是FD&C Act下的化学药。 与FD&C Act介绍部分的末尾划分遥相呼应。
读完第二章的开篇部分,作者想要传达的核心逻辑已经很清晰:美国对生物制药的监管不是“从零开始设计的”,而是在两部法律、两个中心、两条路径的拼图中不断调适过来的。 这个框架在2020年才完成最近一次重大归位,而对于第四波的基因治疗产品,归类逻辑仍在细化中。
站在CMC从业者的视角,理解这个双法源结构至少有三层实际意义:
看懂产品审评路径:为什么同一个生物药在过去和现在走的路径不同?原因可能不在产品本身,而在法律框架的变迁
制定CMC策略时能预判审评预期:读懂CDER和CBER对CMC的审评风格差异,确定产品归到哪个中心管,Reviewer关注的侧重点有哪些不同
在做合规判断时知道从哪个层级的文件中找寻答案:CFR是法律底线,GFI是最佳路径,FR是风向标
下一期我们继续拆解第二章的后续部分:FD&C和PHS两条法律的相通之处,以及在上市后的CMC监管上存在的三个至今仍然有效的实质性差异,即FDA的商业批次放行权、标签后身份检测的法规要求,以及美国独有的“四位字母”生物药后缀制度。这些差异对生物制品商业化后的CMC操作有直接的约束力,也是很多非美国企业在准备FDA申报时容易忽略的地方。
本号所有内容仅为个人学习心得与行业技术交流,不构成任何专业建议,本号会尽力确保内容的准确和完整,若存在理解不当之处,您可在公众号对话框或评论区中发送反馈。
本文所引摘自《The Challenge of CMC Regulatory Compliance for Biopharmaceuticals》第四版,版权归原作者及出版社所有,此处仅供非商业学习交流。