2025年12月25日,国家药监局发布2025年第126号公告,《医疗器械出口销售证明管理规定》将于2026年5月1日起正式施行。
这次的新规发布是证明分类、有效期管理、监管机制等层面都有实质性调整。
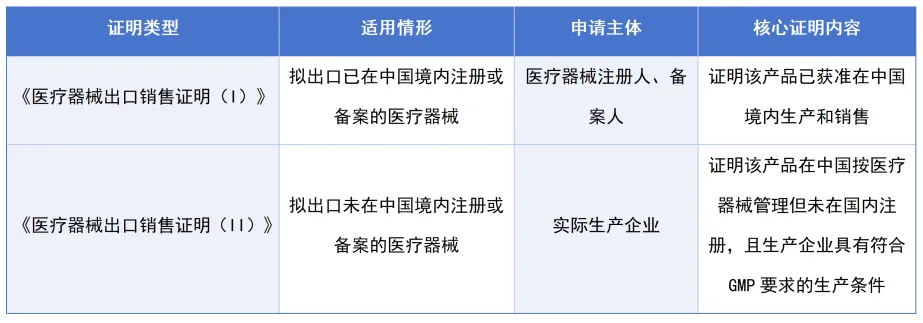
这是新规最重要的结构变化之一。根据产品是否已在中国注册/备案,将出口证明分为两类:
证明(II)的设立是最大亮点,专门针对"海外市场先行、国内注册滞后"的创新产品。
典型场景是:企业的创新器械已获得美国FDA突破性设备认定或欧盟MDR认证,但国内注册流程尚未完成。这种情况下,企业可以凭证明(II)在不等待国内审批的情况下合法出口。
需要注意的是,证明(II)只确认生产条件合规,产品能否进入目标市场,仍取决于进口国(地区)的法规要求。
新规明确:证明有效日期不应超过申报资料中企业提交的各类证件最先到达的截止日期(如注册证到期日、生产许可证到期日等)。第一类医疗器械的出口销售证明有效期不超过3年。
这一调整更务实。以往“一刀切”的2年上限,对那些注册证或生产许可证有效期较长的企业反而是一种限制。新规与证件实际有效期挂钩,避免了“证明还在、证件已过期”的尴尬。
同时,当证明载明内容发生变化,或相关证件被吊销、撤销、注销时,证明应当自相应事项发生之日起失效——这也强化了动态管理的要求。
新规新增/明确了几个硬性要求:
1. 现场检查权
出具证明的部门应当对申报资料的真实性、合法性、有效性以及生产企业的生产质量管理规范符合性进行审核,必要时可以开展现场检查。综合评价符合GMP要求的,予以出具;不符合的,不予出具。
2. 不予出具的具体情形
新规明确列出三种不予出具的情形:
• 列入市场监督管理严重违法失信名单
• 提供虚假资料
• 违反医疗器械监督管理相关规定,处于责令停产整改、涉案处理期间
3. 骗取证明的严厉惩罚
通过虚假资料或欺骗手段骗取出口销售证明的,公示作废并在信用档案记录,5年内不再为其出具出口销售证明;涉嫌违法犯罪的,移交相关部门处理。
4. 追溯义务
申请人应当建立并保存质量管理文件、生产记录、出口销售证明、包装标签样式、报关单、数量、产值、进口国家(地区)等相关资料,保证医疗器械出口过程可追溯。
新规还释放了一些便利化信号:鼓励省级药监部门实行网上办理,推动全流程电子化。
内容来源: 国家药监局2025年第126号公告《医疗器械出口销售证明管理规定》