免责声明:本文仅为群友学习心得,不构成任何医疗建议.
我们的平生群共同完成了《肿瘤生物学》第一季的学习。群友们在陆续记录自己的学习心得,特别是从这些基础知识引发的与自己治疗实际相结合的思考.
此刻阅读的你,也许是一个群外的伙伴,也许是未来的某一个新患,我们这群人原本也和你一样是与医学无关的人,但我们用我们的努力的去理解医学,去驱散恐惧。
小南说明:
今天选发这篇学习笔记的作者是群里一个叫“石头”的姑娘,她家是胆道患者,从一个小白家属学起,在和大伙一起学习”肿瘤生物学”教材的同时举一反三的去联想到自己家的治疗实际.写的这份笔记也引用了参考文献.
发表前,我也曾犹豫是不是要做点修改,毕竟这是公众平台。但我又想到自己也是从一个小白零基础学起,当前写下的理解和分析可能还存在偏颇,参考的文献质量也有待提高,但医学本身就是在不断质疑和修正中进步的。何况我们。因此,我们决定保留现阶段的学习记录,不急于修改。
本文并非是给予公众任何医学建议,而是记录我们的学习态度。我们学习中的所思所考,最终都是要与医生探讨。我们此时的认知也许会在彼时被更正。但,这是一个动态成长的过程——我们会坚持学习,持续更新知识体系,正如医学在持续探索中向前发展一样。
学习不是为了给自己开药,而是为了在医生面前做个明白人。
为什么学习肿瘤生物学?
文 | 石头
在群里,跟着病友们一起学习肿瘤生物学,最大的收获不是记住了几个生涩的医学名词,而是完成从“盲目恐慌”到“建立体系化策略思维”的认知转变。
面对晚期恶性肿瘤,过去常常将其视作一个固定不变的细胞团,从而将治疗简化为“见瘤打瘤”的局部对抗。但通过系统学习,我了解到:肿瘤是一个具有高度适应性、在持续生存压力(药物、缺氧、免疫监视)下不断进行克隆竞争与演化的动态的微型生态系统。
如果治疗策略的认知维度低于肿瘤的演化维度,临床干预将永远滞后于其耐药机制。
学习肿瘤生物学,旨在建立全局视角。
这意味着治疗的终极目的不仅是追求影像学上病灶体积的缩小(这往往只是清除了敏感克隆,却通过竞争释放为耐药克隆腾出了位置,更在于如何通过干预病人代谢通路、重塑免疫微环境,营造一种打破癌细胞稳态的生态。只有深度解析肿瘤的基因情况(驱动力)、代谢偏好(燃料)与微环境屏障(防御),才能有效调动人体适应性免疫系统对其进行精准的合围与剿灭。
一、 基因层面
肿瘤的演化遵循达尔文进化论,厘清其结构是实施精准打击的前提。
一方面:
主干突变存在于几乎所有肿瘤细胞中,其VAF理论上应接近肿瘤纯度×0.5(杂合)或1.0(纯合),它们在肿瘤演化的极早期出现。在临床意义上,这是一个理论上疗效可靠的治疗靶点。 靶向主干突变,其打击面覆盖整个肿瘤,不易因空间异质性导致逃逸。
然而,在实际临床 NGS 报告中,变异等位基因频率(VAF)受基因杂合性、拷贝数变异(CNV)以及肿瘤纯度三大变量联合影响。如在致密促纤维增生型肿瘤(如 ICC)中,由于含有大量的成纤维细胞和免疫细胞(间质污染),实际 VAF 会被显著稀释。因此,仅凭 VAF 判断克隆性存在局限。
此外,部分肿瘤呈现非线性、平行演化(多中心起源或早期分支),这使得单纯靶向某一主干突变的策略可能失效。
分支突变(亚克隆突变)仅存在于部分细胞群中,通常在治疗压力下富集或后期演化中产生。单独靶向分支突变无法消灭整个肿瘤。且高肿瘤异质性意味着肿瘤内部“门派林立”,预示着更差的预后。
另一方面:
某些突变在赋予癌细胞生长优势的同时,也破坏了其冗余备份机制,使其在进化路径上被“锁定”。
以我们家基因情况为例,ARID1A + PBRM1 双突变导致 SWI/SNF 染色质重塑复合物功能丧失,加重了基因组不稳定性(增殖优势),但也损害了 DNA 双链断裂修复中的同源重组通路。癌细胞因此被迫高度依赖备用的 ATR-CHK1-WEE1 轴来维持 G2/M 细胞周期检查点。此时引入 ATR 抑制剂或 WEE1 抑制剂,可迫使带有 DNA 损伤的癌细胞提前进入有丝分裂,引发有丝分裂灾难而凋亡
二、代谢层面
肿瘤代谢具有高度的组织特异性与可塑性。
1. 胆汁酸代谢紊乱与炎性癌变
蒙南哥提醒,我们特别关注了胆汁酸肝肠循环方面的问题。多项基于人群的队列和病例对照研究发现,胆石病与肝内胆管癌(iCCA)风险显著相关,OR值在2.0–5.8之间,且风险随结石体积和病程延长而增加。胆囊切除术本身是否独立增加风险尚存争议,可能与术后胆汁持续流入、次级胆汁酸积累有关。主流机制学说包括:
a) 慢性激活学说:造成长期机械损伤+胆汁淤积→胆管上皮损伤、修复→DNA损伤累积→癌变(即 “炎性癌变”路径,学界称“慢性炎症诱发癌变”)
b) 胆汁酸激活诱发学说:胆囊作为胆汁的“储存与浓缩器官”被切除后,胆汁持续排入破坏,引起肠肝循环改变、次级胆汁酸(如脱氧DCA、石胆酸LCA)比例上升,这些次级胆汁酸具有损害细胞毒性和促炎、促癌的作用
c) 破坏菌群-胆汁酸平衡舒缓:肠道菌群通过7α-脱羟基酶将初级胆汁酸(如CA、CDCA)转化为次级胆汁酸(如DCA、LCA)。胆道梗阻或胆囊切除后,胆汁酸谱改变,促进促炎菌群扩增,形成“菌群-胆汁酸-炎症”恶性循环,加剧胆管上皮损伤。
2. 胆汁酸驱动免疫逃逸
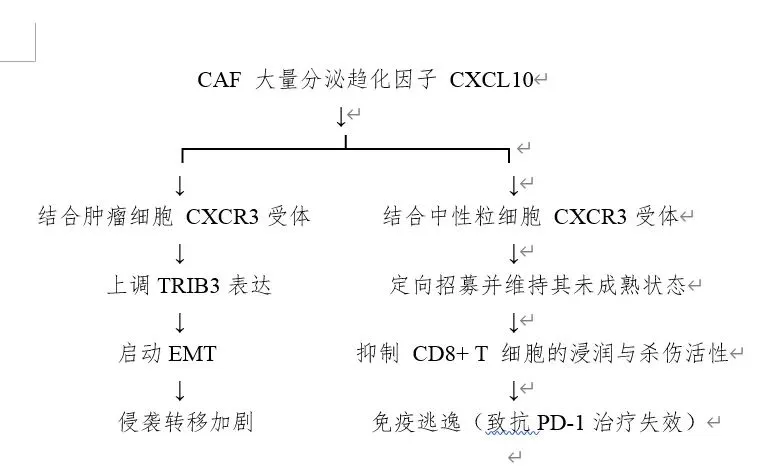
近年研究揭示,胆汁酸不仅是代谢副产物,更是细胞间通讯信号分子,山东大学齐鲁医院团队在 Cancer Cell 上的研究系统阐明了胆汁酸如何通过癌相关成纤维细胞间接驱动免疫逃逸。
3. 代谢“搭便车”效应:
转移灶的代谢模式呈现高度的生态位适应。例如:淋巴结转移灶可能重编程为依赖脂肪酸氧化(FAO);而腹膜转移灶由于极度缺血缺氧,肿瘤细胞会诱导周围的 CAFs 进行有氧糖酵解产生乳酸和脂肪酸,肿瘤细胞则摄取这些代谢废料进行氧化磷酸化(OXPHOS)供能,即“反向瓦伯格效应”。
三、 肿瘤微环境层面
根据研究,ARID1A/PBRM1缺失不仅改变细胞内基因表达,还通过以下机制促进免疫逃逸:
1. 抑制I型干扰素通路
SWI/SNF 正常时负责维持 cGAS-STING 信号通路的染色质开放。ARID1A 缺失导致 STING 启动子区染色质致密化,转录受抑;阻断 I 型干扰素释放,导致树突状细胞(DC)无法提呈抗原,T 细胞招募失败(肿瘤变“冷”)。
2. 表观遗传驱动 PD-L1: ARID1A 缺失导致染色质空间构象异常,从而影响转录因子及共激活因子在 PD-L1 启动子/增强子区域的结合,或促使组蛋白修饰异常(如 H3K27ac 改变),形成独立于传统 IFN-γ 诱导的表观遗传驱动型 PD-L1异常表达。
3. 富集免疫抑制细胞:促使 Treg(调节性 T 细胞)和 MDSC(髓系抑制细胞)大量浸润,形成坚固的免疫抑制屏障。
四、 免疫层面
化疗是无差别杀伤增殖细胞,易诱发耐药克隆的竞争释放;靶向药精准打击特定靶点,但不可避免地驱动旁路激活。而适应性免疫系统凭借其 T 细胞受体库的高度多样性、克隆扩增的动态适应能力及长期免疫记忆,是唯一能在时间维度上跟上肿瘤演化速度的武器。
然而,有效的抗肿瘤免疫必须完成经典的“肿瘤-免疫循环”。iCCA 极其致密的促结缔组织增生微环境,在这个循环的多个关键节点设置了重重障碍:
节点一:抗原提呈受阻
肿瘤微环境大量分泌VEGF、IL-10 以及核心免疫抑制因子 TGF-β。这一细胞因子网络不仅促进异常血管生成,还直接阻断单核细胞向树突状细胞(DC)分化,冻结 DC 细胞的成熟过程,阻断抗原向淋巴结的递送。
节点二:血管壁外渗困难
iCCA 内部大量畸形的异常血管网络,叠加致密细胞外基质,导致组织间质流体压力升高。这种高压状态及内皮细胞黏附分子的下调,使得血液中的效应 T 细胞无法黏附并穿透血管壁进入肿瘤基质。
节点三:基质排斥与代谢耗竭
即使极少数 T 细胞艰难渗出,也会遭遇由成纤维细胞(CAF)和致密 I 型胶原网络构筑的物理壁垒(即“免疫排斥表型”),被卡在肿瘤实质外围。同时,肿瘤核心区的严重缺氧、酸中毒(乳酸堆积)以及色氨酸耗竭(IDO 酶过度表达),会迅速引发浸润 T 细胞的代谢耗竭,上调 PD-1/TIM-3/LAG-3 等耗竭标志物,彻底丧失细胞毒性杀伤功能。
这也就是为什么南哥分享中时常提到,对于成纤维细胞富集+乏血供的实体瘤,单靠 PD-1/PD-L1 抑制剂往往不够,而需进一步考虑联合化疗(诱导免疫原性细胞死亡 ICD 释放抗原)、抗血管生成药物(重塑畸形血管降低 IFP),或靶向代谢/基质药物(瓦解 CAF 墙),重塑微环境,才能真正释放免疫治疗的潜能。
(文完)
一位抗癌11年群主致患友们的一封倡议信
肿瘤生物学1:迈出成为学习型家属的第一步
肿瘤生物学2:人为什么会得癌症?
肿瘤生物学3.单细胞测序视野下的肿瘤
肿瘤生物学4.基因测序
肿瘤生物学5. 腺癌、鳞癌和其他组织来源癌
肿瘤生物学6.同病异治的原因
肿瘤生物学7. 不同来源癌的治疗逻辑
肿瘤生物学8:白血病/淋巴瘤和神经系统肿瘤等
肿瘤生物学9.实体瘤可以从血液瘤中借鉴什么?
肿瘤生物学10. 同源重组修复和错配修复
肿瘤生物学11:遗传和环境
肿瘤生物学12.家有患者其他正常人要注意什么?
肿瘤生物学13.病理诊断的更多价值发掘
肿瘤生物学14.肿瘤抗原
肿瘤生物学15.抗原如何利用?
肿瘤生物学16.原发灶与转移灶
肿瘤生物学17.微环境中的Tregs及Ctla4等
肿瘤生物学18. 增生- 化生-不典型增生-原位癌
肿瘤生物学19.癌细胞逆转正常细胞
在知识中寻找力量
学习不是要成为专家,而是让陪伴更有质量,让选择更有依据。愿我们在这条路上继续彼此支撑,用理解照亮前路。
—— 小南