上篇讲了慢病毒种属特性及其生命周期,本篇重点讲述慢病毒的科研及临床应用,提供可选的研发方向与思路,不提供具体改造方案。
内容包括:
慢病毒载体生产系统
慢病毒假型化包膜蛋白
病毒融合蛋白分类
慢病毒假型化机制
慢病毒载体靶向重定向
结语
说明:可用于慢病毒假型化的包膜蛋白以病毒科属进行分类,并列出包膜蛋白所来源的病毒种属、嗜性、该假型化病毒特性;除弹状病毒科与逆转录病毒科内参考文献未完全确认外,其他病毒都已经完成查证,其中前30条参考文献由AI查询得来,检查的时候发现适配性存疑,又人工补了后续的文献;前几天我的小狗豆豆得了细小,每天带着去输液,实在没心情查,但这篇文章实在拖得太久就先发出来,参考文献的查证对文章影响有限,可以先参考阅读,我补全后更新在‘阅读原文’的链接里。
有什么错漏请留言并请包涵!
慢病毒载体生产

野生型慢病毒可以经过改造为在实验环境中安全使用的慢病毒载体,这些改造的慢病毒载体具有多种优点,成为现今多种细胞和模型中有力的研究工具。慢病毒载体可靶向非分裂细胞,可在宿主长期稳定表达,并且表现出低免疫原性。
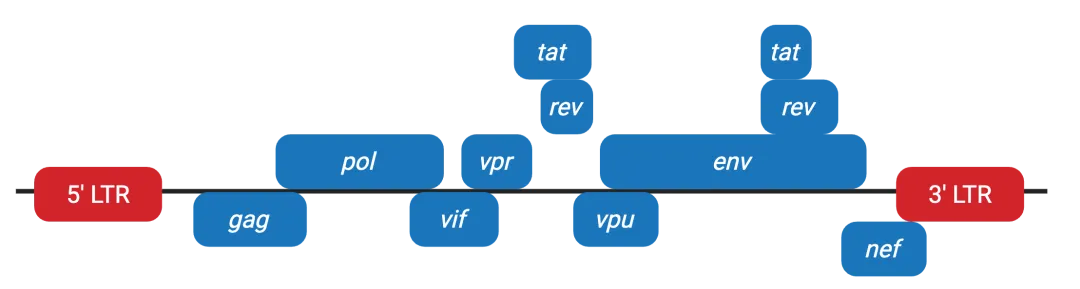
图1. 野生型慢病毒基因组
慢病毒的单链RNA基因组包含包装基因、调节基因、辅助基因以及整合所需的LTRs,所有慢病毒均使用包装基因gag、pol、env来制造病毒颗粒,野生型慢病毒额外需要调节基因tat和rev,以及病毒特异的辅助基因(如HIV-1的vif、vpr、vpu和nef)。LTR包夹所有基因,其中的任何序列都会被整合进宿主基因组。
然而,并非所有组件都是慢病毒生产必需的,许多非必需组件被移除或突变,而需要的组件则被分割到不同的质粒中,以此增加系统安全性。
为制备慢病毒载体,你需要3个(或第三代的4个)质粒:
穿梭质粒:包含目的基因、sgRNA或shRNA,两侧被LTR序列包夹,包装容量8-10kb
包装质粒:包含包装基因gag和pol,以及调节基因tat和rev,在第三代系统中被分割到2个质粒中
包膜质粒:包含包装基因env(通常使用具有广谱感染性的VSV-G)
当前我们所使用的慢病毒载体递送工具,主要是由HIV-1改造而来,经过多年的改进提升了效率和安全性,这些改进将慢病毒载体划分为三个代次。
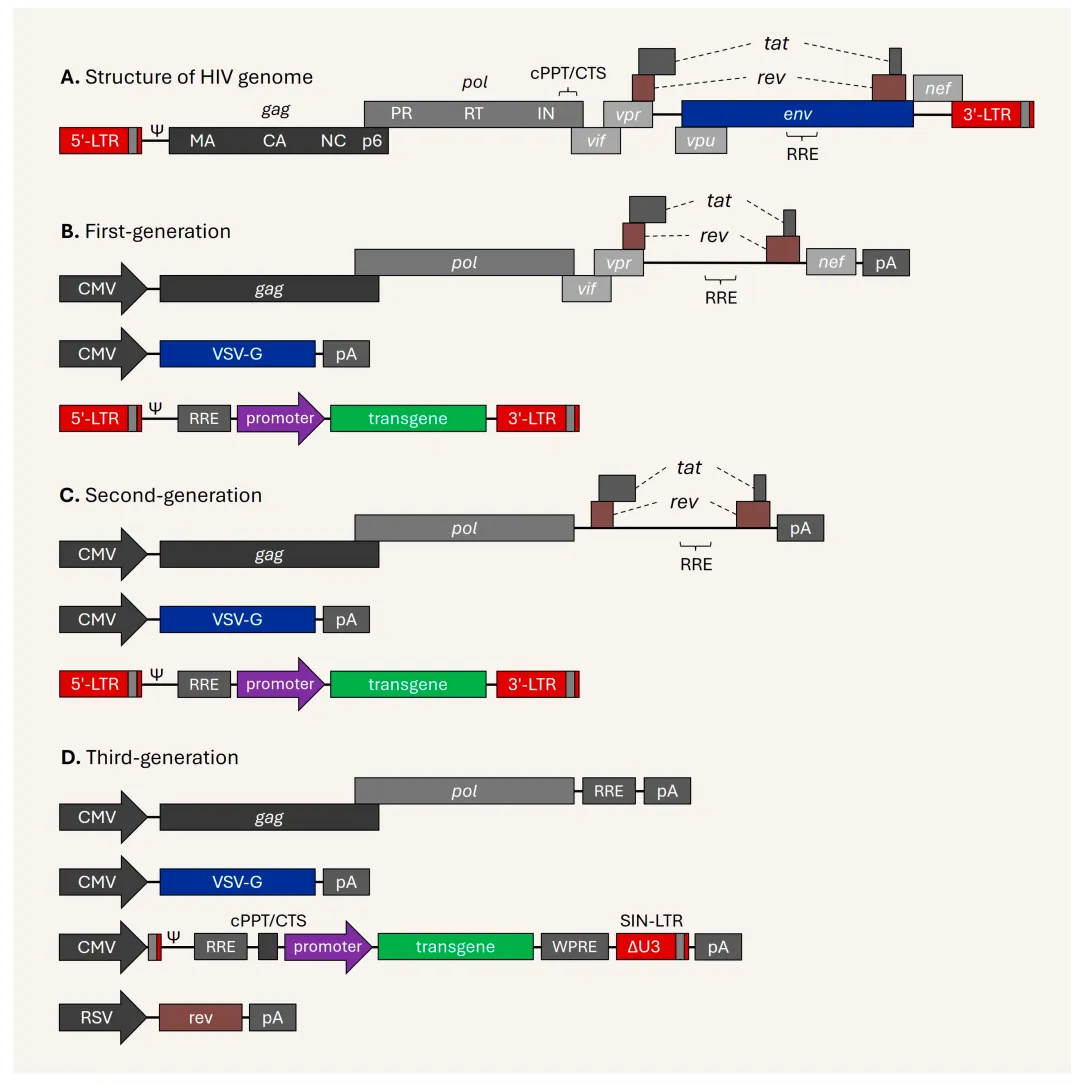
图2. HIV基因组及三代慢病毒载体示意图
1. 第一代
第一代慢病毒质粒中的病毒组件虽然被分离到不同的质粒中,但病毒基因组基本保持完整,如图2B。由于缺乏安全性,且可能产生具有复制能力的慢病毒,目前已基本弃用。
第一代质粒包括:
2. 第二代
第二代慢病毒质粒通过移除辅助基因极大地提高了慢病毒载体生产的安全性,但保留tat基因,因为穿梭质粒5' LTR的启动子活性需要Tat蛋白来激活。如图2C。
第二代质粒包括:
穿梭质粒:包含目的基因和野生型LTRs
包装质粒:包含gag、pol、tat和rev
包膜质粒:包含env
3. 第三代
第三代系统进一步提高了二代系统的安全性。首先,包装系统被拆分为2个质粒,虽然更加安全,但使用起来更加复杂,额外的质粒也造成病毒滴度更低;其次,三代系统不再需要tat,穿梭质粒包含一个与异源启动子(通常是CMV或RSV)嵌合的5' LTR,从而无需Tat蛋白的转录激活。
大多数第三代穿梭质粒的3' LTR区域存在缺失,使其具有自失活 (self-inactivating, SIN) 能力。该缺失经过逆转录后转移到5' LTR区域,从而抑制病毒整合到宿主基因组后全长病毒的转录。该设计在第二代穿梭质粒中也较为常见。见图2D。
第三代质粒包括:
需注意,三代穿梭质粒可以使用二代或三代包装质粒进行包装,而二代穿梭质粒只能使用二代包装质粒进行包装,二代系统与三代系统的差异汇总如下表:
| | |
| 只能被含有tat基因的二代包装系统包装 | 二代和三代包装系统均可包装 |
| 一个质粒,编码gag、pol、tat和rev | 二个质粒,一个编码gag/pol,另一个编码rev |
| 可互换,通常编码VSV-G | 可互换,通常编码VSV-G |
| 安全;通过三个独立质粒编码必要的病毒基因,获得复制缺陷的病毒 | 更安全;通过四个质粒实现复制缺陷且无需使用Tat蛋白。含3' LTR缺失实现自失活 |
| | 杂合启动子;5' LTR 部分缺失并与异源增强子/启动子(如 CMV 或 RSV)融合 |
慢病毒载体制备:慢病毒生产通常使用人胚肾293T (HEK293T) 细胞作为生产细胞,将所需穿梭质粒、包装质粒与包膜质粒转入HEK293T细胞后,细胞将组装病毒颗粒并将其释放到培养基上清中。质粒转染经过首次换液后,可收集含病毒的培养上清储存或进行离心浓缩,无论是否检测病毒滴度,都可以使用粗提或浓缩病毒进行靶细胞转染。
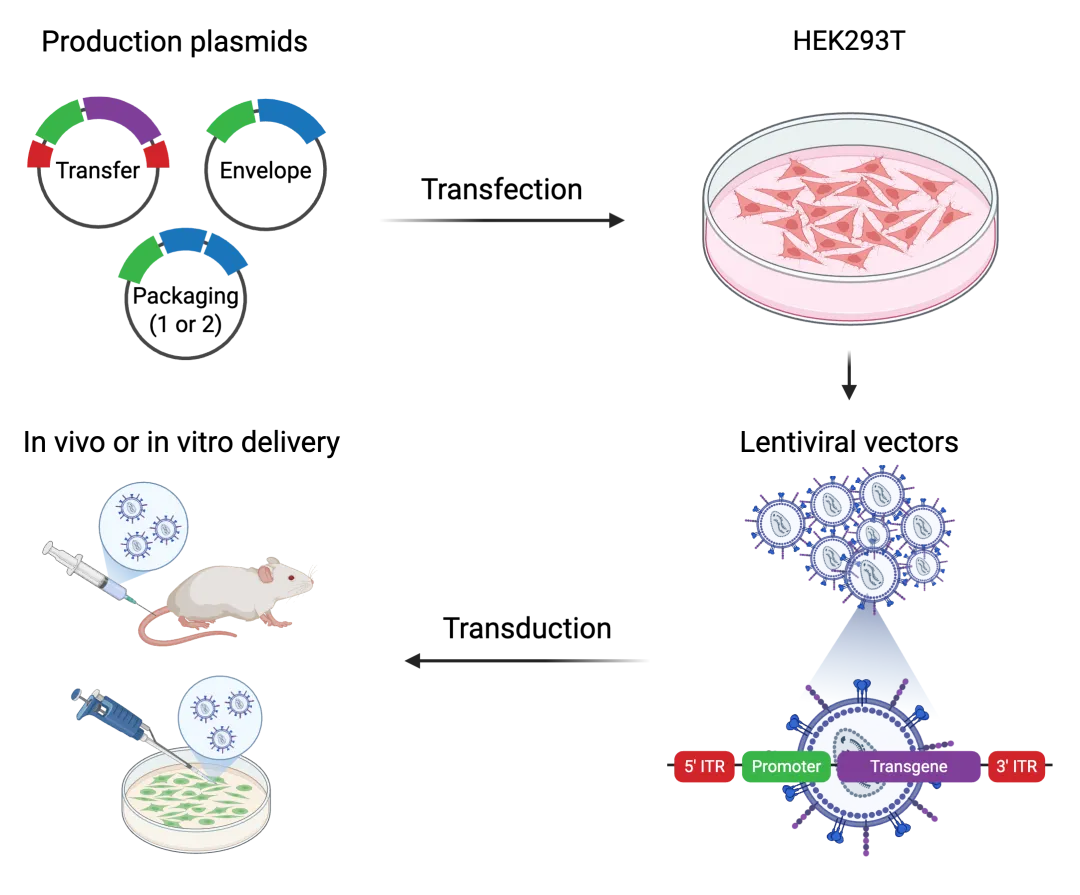
图3. 从生产质粒开始的慢病毒载体生产示意图
由于慢病毒转染细胞后会整合进入基因组,有可能造成未知风险,且某些使用场景只需瞬时表达,这时就可以使用整合酶缺陷型的慢病毒载体(integrase-deficient lentiviruses, IDLVs),与野生型慢病毒相比,IDLVs将基因组RNA反转录为DNA后,由于整合酶失活,而线性前病毒DNA已准备好整合,因此可以通过同源重组或非同源末端连接产生两种类型的环状附加体(episome)。
这些附加体缺乏复制起点,无法在分裂细胞中维持,但IDLV s基因组具有转录活性,可支持基因产物的瞬时表达,随着时间的推移,附加体在细胞分裂周期中丢失,基因表达逐渐减弱,但丢失速率很大程度上取决于细胞的更新速度。
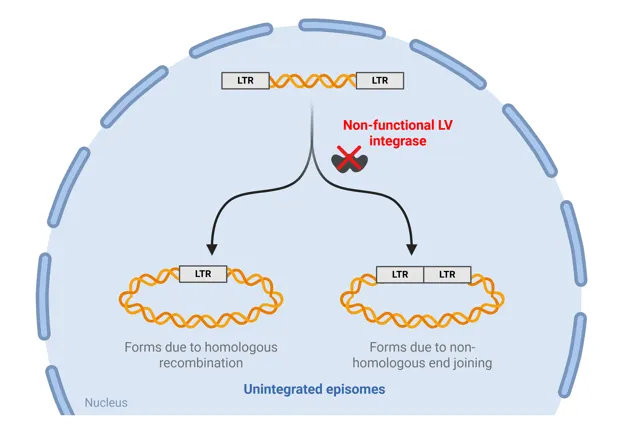
图4. 非整合慢病毒的整合过程与附加体形成示意图
贰
慢病毒载体假型化

嗜性决定了慢病毒可以感染的细胞类型,如HIV-1的包膜蛋白gp120/gp41可结合T细胞表面CD4受体与辅助受体,从而感染CD4+ T细胞,但通过更改包膜基因可改变病毒嗜性,这一过程称为假型化。例如目前最常用的慢病毒假型化包膜蛋白VSV-G,其受体为多种细胞表达的LDLR,VSV-G假型化的慢病毒可实现多种类型细胞的高效转染。另外,假型化还可以更安全地研究未知的病毒病原体,例如新冠疫情期间,使用COVID-19的Spike蛋白假型化慢病毒,模拟研究新冠病毒的感染机㓡。此外,假型化还可以降低病毒毒性,或改善病毒对血清的敏感性。
基于已开展的研究成果,慢病毒可成功假型化的包膜蛋白按病毒科分类如下:
1. 弹状病毒科Rhabdoviridae
Envelope Protein | | | | |
| Vesicular stomatitis virus | LDL-R (Low-density lipoprotein receptor) | Broad spectrum (most mammalian cells) | 金标准,高滴度、高稳定性,可被血清补体灭活,对静息免疫细胞效率低 |
| | Neuronal cell surface receptors (nAchR, p75NTR, NCAM) | | |
| | LRP1 (presumed, neural cell receptor) | | |
| | Presumed LDL receptor family member | | |
| | Presumed LDL receptor family member | | |
| | Presumed LDL receptor family member | Hematopoietic stem cells, T cells | |
| | | | |
2. 逆转录病毒科Retroviridae
| | | | |
| Human immunodeficiency virus | | CD4⁺ T cells, macrophages | |
| Murine leukemia virus (amphotropic 4070A) | | | |
| Murine Leukemia virus (amphotropic 10A1) | | | |
| Gibbon Ape Leukemia virus | | | |
| Feline endogenous retrovirus | | CD34⁺ hematopoietic stem cells, T/B cells | 非细胞毒性,转导效率优于VSV-G,血清稳定性增加 |
| Baboon endogenous retrovirus | ASCT1 (SLC1A4) / ASCT2 (SLC1A5) | Resting HSPCs and T, B, NK cells | 在静息造血细胞转导方面显著优于VSV-G和HERV-W,可利用双受体进入 |
| | THTR1 (Thiamine transporter 1, KoRV-J subtype) | Freshly isolated immune cells | |
| Jaagsiekte sheep retrovirus | | | |
| Avian sarcoma leukosis virus | TVA (A subgroups) / TVB (B/D/E subgroups)/TVC (C subgroups) | | 无背景感染,可工程改造实现抗体介导再靶向,高热稳定性,低免疫原性 |
3. 副黏病毒科Paramyxoviridae
| | | | |
| | | | 需共表达H(血凝素)和F(融合蛋白),滴度较低但靶向精准,高既有免疫人群 |
| | | | |
| Human Parainfluenza virus | | | |
| | | EphrinB2⁺ cells,包括内皮细胞、神经细胞与干细胞等 | |
4. 肺病毒科Pneumoviridae
| | | | |
| Human Respiratory Syncytial virus | heparin,CX3CR1,nucleolin,ICAM-1 | | |
5. 杆状病毒科Baculoviridae
| | | | |
| Autographa californica multiple nucleopolyhedrovirus | | | |
6. 肝病毒科Hepadnaviridae
| | | | |
| | NTCP (Sodium taurocholate cotransporting polypeptide) | | |
7. 丝状病毒科Filoviridae
| | | | |
| | | 广谱,感染单核、巨噬与DCs,以及内皮细胞、肝细胞和肾上腺皮质细胞 | 高度糖基化形成糖链屏障,隐藏抗原表位;需要蛋白水解去除糖基帽,暴露结合位点 |
| | | 广谱,可感染巨噬细胞、单核细胞与DCs,内皮细胞,具有肝嗜性 | 丝状病毒研究工具,通过巨胞饮作用进入细胞,侵染依赖于组织蛋白酶 |
8. 披膜病毒科Togaviridae
| | | | |
| | | | |
| | Mxra8, PHB1, GAGs and TIM-1 | 广谱,感染关节、皮肤、肌肉和肝脏,可感染肌肉、成纤维和滑膜细胞系等 | |
| | | | |
| | | | |
9. 正黏病毒科Orthomyxoviridae
| | | | |
| | sialyl-galactosyl residues | | 需NA和M2蛋白辅助生产,蛋白水解激活,pH依赖性融合 |
| | | | |
10. 呼肠孤病毒科Sedoreoviridae
| | | | |
| | sialic acids/ HBGA+Integrins/Hsc70/JAM-A &Occludin | | |
11. 沙粒病毒科Arenaviridae
| | | | |
| Lymphocytic Choriomeningitis virus | | | |
| | | 广谱嗜性,可高效转染内皮细胞、成纤维细胞与上皮细胞细胞系,可感染APC | |
对慢病毒进行假型化使用的包膜蛋白具有两个核心功能:提供特异性靶向结合能力,从而改变慢病毒嗜性;提供膜融合能力,实现病毒载体的细胞内递送。对于不同的病毒来说,这两个功能可由单一蛋白完成如VSV-G,也可由两个或多个不同的蛋白分别执行不同的功能如Measles H/F。
叁
病毒融合蛋白分类

根据结构特征,病毒膜融合蛋白(membrane fusion proteins)可分为三类,不同融合蛋白还包含不同类型的融合肽,并且对辅助蛋白的依赖程度也各不相同。尽管如此,所有已鉴定的病毒融合蛋白都会经历一个过程:首先从具有融合能力的状态(二聚体或三聚体)转变为嵌入膜内的同源三聚体前发夹结构,然后转变为发夹三聚体,该发夹三聚体使附着于靶膜的融合肽和附着于病毒膜的跨膜结构哉紧密靠近,从而促进病毒膜和靶膜的融合。在这些构象转变过程中,融合蛋白诱导膜结构经历紧密贴合、半融合、形成小融合孔以及最终形成大融合孔等阶段。大量研究表明,不同病毒融合蛋白都趋向于采用相同的总体策略来介导融合,然而这些融合蛋白在序列和结构上却差异很大,并且由不同的触发机制激活。
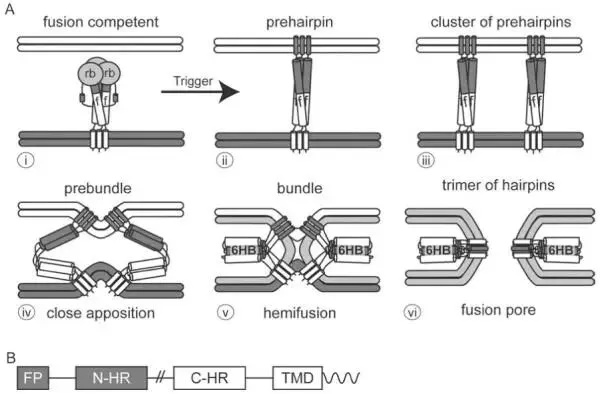
图5. 常见三聚体发夹结构的膜融合途径
I类融合蛋白
I类融合蛋白在融合前和融合后均为三聚体,其最终状态的典型特征是具有一个中央N端三聚体α螺旋卷曲螺旋,并由三个‘C端’螺旋修饰,从而形成一个六氢化物链(6HB)。I类蛋白的6HB大小和位置差异显著,它们的融合肽位于融合亚基的N端或附近。
II类融合蛋白
II类融合蛋白主要由β折叠结构构成,并排列成3个相连的结构域,其排列方向与膜平行,这与I类病毒膜融合蛋白有显著差异,其内部融合肽以环状结构位于β链末端。它们与伴侣蛋白结合,该伴侣蛋白在病毒组装过程中或组装后不久被切割。成熟后,融合蛋白的胞外结构域以反平行二聚体的形式存在于病毒体表面下方,每个茎部位于病毒体的三重对称轴上,一旦被激活,II类融合蛋白会重新排列成三聚体,并从病毒膜的三重对称轴上突出。
III类融合蛋白
III类融合蛋白兼具I类和II类融合蛋白的特征,与I类融合蛋白类似,它们在融合前均为三聚体,并包含一个中央α螺旋卷曲螺旋结构,但是它们的融合结构域更类似于II类融合蛋白,其融合环位于延伸的β折叠链的末端。以HSV-1 gB和VSV G为代表,该类蛋白的外结构域可分为5个亚结构域,其中结构域I被称为融合结构域,主要由β折叠构成,顶端包含融合环,与第二类膜融合蛋白相同
肆
异源病毒包膜蛋白假型化机制及限制条件

一、异源包膜蛋白能嵌入的核心机制
慢病毒包膜的假型化本质上是脂质膜蛋白的物理整合过程,而非严格的生物识别机制。其可靠性基于以下原理:
1. 病毒出芽的膜来源特性
慢病毒颗粒的包膜直接来源于宿主细胞质膜的脂筏区域,这些区域富含胆固醇和免疫受体。任何嵌入细胞膜的异源糖蛋白,只要位于脂筏附近,就有概率被出芽的病毒颗粒捕获并包裹进去,这一过程不需要慢病毒与异源包膜蛋白之间的特异性识别。许多异源病毒(如丝状病毒或弹状病毒)具有与慢病毒类似的组装机制,使其同源三聚体结构包膜糖蛋白能够有效地与慢病毒核心结合。
2. Gag蛋白的招募作用
病毒结构蛋白Gag在质膜组装时,其基质蛋白(MA)结构域会与膜脂质及嵌入的跨膜蛋白胞质尾部相互作用。这种招募机制不依赖包膜蛋白的序列特异性,而是依赖其跨膜区和胞内区的拓扑结构。只要异源蛋白具有合适的跨膜锚定结构,就能被Gag招募进病毒颗粒。
3. 翻译后修饰的兼容性
酰化等脂质修饰可增强包膜蛋白对脂筏的亲和力,显著提升假型化效率。多数病毒的包膜蛋白本身就具有这类修饰信号,使其能自然定位到病毒出芽位点。
二、无法进行慢病毒假型化的包膜蛋白类型
尽管慢病毒假型化具有广泛兼容性,但以下类型的包膜蛋白存在根本性限制:
1. 结构缺陷型蛋白
2. 需要病毒特异性辅助因子的蛋白
某些病毒的包膜蛋白需要其同源病毒特有的辅助蛋白才能完成正确折叠或运输。例如:
需要特定伴侣蛋白进行构象成熟的包膜蛋白
依赖病毒编码的蛋白酶进行剪切激活的融合蛋白
在缺乏辅助因子的异源系统中无法形成功能性构象
3. 结构过度复杂或不兼容的蛋白
4. 诱导强烈细胞毒性或干扰机制的蛋白
5. 缺乏广谱受体识别能力的蛋白
三、影响假型化效率的关键因素
1. 胞内尾区的兼容性
包膜蛋白的胞质尾区长度和序列显著影响其被Gag招募的效率。过长的尾区可能空间位阻Gag互作,而某些病毒的尾区含有特殊的晚期结构域 (late domain), 可促进出芽。
2. 脂筏定位信号
包膜蛋白是否富含胆固醇结合基序或棕榈酰化位点,决定其能否有效富集于病毒出芽位点。
3. 生产系统的适配性
无法进行慢病毒假型化的包膜蛋白类型,需要进行适当调整或修饰以满足膜嵌入机制。在慢病毒的假型化过程中,如果外源蛋白的膜内结构过于庞大或形状怪异,会与正在形成的Gag蛋白晶格发生碰撞,将导致病毒在没有“外皮”的情况下出芽,产生一种“光秃秃的”、不具感染性的病毒颗粒。
对于无法整合的外源蛋白,我们通常进行尾部替换,即将目标病毒的“头部”(胞外结构域)与VSV-G或HIV-1 gp41的“尾部”(跨膜区和胞质区)融合。这样就形成了一种嵌合蛋白,其外部结构与目标病毒相似,但内部功能却与慢病毒相容。
伍
病毒载体靶向重定向

在in vivo细胞疗法场景中,需要在保留包膜蛋白融合能力的同时对其原有嗜性进行工程改造,使慢病毒载体特异性地靶向目的细胞,以避免体内应用时病毒载体脱靶感染非目的细胞造成未知风险。
为此,通常采用‘blind and bind'的策略对包膜蛋白进行工程化,以VSV-G为例:
1. ‘致盲’VSV-G:引入突变使VSV-G无法结合其天然LDLR受体,失去原有的组织或细胞嗜性但保留膜融合能力,通过结构工程已证实关键突变如K47Q和R354Q可有效致盲VSV-G。
2. 重定向靶向:盲化后,需将新的靶向机制添加到病毒包膜,其中主要有两种主要方法
在体内CAR-T生产中,可以使用anti-CD3或anti-CD7序列对载体进行改造,从而直接在患者体内制造CAR-T细胞,无需昂贵的以及长周期的生产过程。
而以麻疹病毒双包膜蛋白H与F蛋白为例,其与单一的 VSV-G 蛋白不同,麻疹病毒包膜采用双蛋白系统:H(血凝素)用于结合,F(融合蛋白)用于进入细胞膜。为了实现体内细胞治疗,需要多步骤的过程来保证载体只感染目的细胞且耐受患者免疫系统:
1. ‘致盲’H蛋白:H蛋白天然结合CD46、SLAM与Nectin-4,为防止载体进入非目标细胞,必须对H蛋白的胞外域进行点突变,常用的突变包括Y481A和R533A,以及S548L和F549S,使其失去与天然受体结合的能力,确保其在生物学上处于惰性状态。
2. 重定向靶向:H蛋白被‘致盲’后,在其C末端融合一个特定的靶向配体,使其能够特异性地识别T细胞表面标记物,如CD3、CD4、CD8或CD7等。
这种方式将结合与融合功能解耦,使靶向配体只负责结合,而不会干扰F蛋白倡导的融合过程。另外,需要注意的是,麻疹病毒属于副黏病毒,其糖蛋白与慢病毒核心在生物物理上并不完全兼容,为使H和F蛋白能高效组装到慢病毒颗粒表面,必须对其N端胞质尾部进行截短,H蛋白通常截短18至24个氨基酸(如CTΔ18或Δ24),F蛋白通常截短30个氨基酸(CTΔ30)。
虽然上述改造可以使VSV-G或H/F蛋白假型化的慢病毒载体重定向靶向T细胞,从而实现in vivo细胞疗法的体内应用,但由于天然VSV-G极易被人类血清中的实体系统灭活,因此还需要进行增强血清抗性的定向进化,或使用与VSV-G高度同源但抗性更强的Cocal-G蛋白 (Umoja Biopharma),以满足全身性给药需求;而麻疹病毒H/F包膜蛋白由于人类普遍搁麻疹疫苗或有感染史,体内存在预存中和抗体限制了其临床应用,因此在实际药物开发时,可使用其他副黏病毒如Nipah病毒糖蛋白G/F (Sana Biotechnology) 进行慢病毒假型化改造。
陆
in vivo细胞疗法——不止慢病毒
无论使用哪种病毒包膜,对包膜蛋白的改造策略基本一致,即”blind and bind“致盲与重定向靶向;此外需要注意的是,由于我们使用的生产细胞株293T自身携带MHC分子,可能会展示于病毒包膜造成免疫清除,因此需要对生产细胞株进行基因编辑敲除MHC分子;而不同生产细胞糖基化的差异,也会造成病毒载体特性的改变;另外,为了避免病毒在体内被吞噬细胞非特异性吞噬,也有必要在病毒包膜表面展示“don't eat me”信号;为了增强体内感染效率,还可以在包膜表面增加相应的活化抗体如anti-CD3/anti-CD28,提高效率降低用量避免高剂量风险。
对于现在的细胞治疗或CGT,甚至整个生物医药行业来说,除慢病毒之外还有更多的病毒有待工程化,使用何种病毒,实现何种目的,满足何种需求,需要我们一起向自然界汲取经验。病毒经历亿万年的演化,是天然进化出的巨大宝库,每个基因或组件都是一个插件,每个病毒基因组都是完整的skill,在工程化过程中需要使用工具时,不妨先向大自然取经。
ps: 由于参考文献比较多,目前是79条,文章中假型化糖蛋白的种类及参考文献单独放在阅读原文链接中,方便补充更新;另外,我使用的是obsidian写的markdown文件,用其他软件打开时可能会造成引文序号错乱,需要注意。
参考资料:
https://www.addgene.org/guides/lentivirus/https://blog.addgene.org/viral-vectors-101-pseudotypingThe Era of Gene Therapy: The Advancement of Lentiviral Vectors and Their Pseudotyping; doi: https://doi.org/10.3390/v17081036https://notebooklm.google.comhttps://www.kimi.comhttps://ictv.global/reportCell type-specific delivery by modular envelope design; doi: https://doi.org/10.1038/s41467-023-40788-8Structures and Mechanisms of Viral Membrane Fusion Proteins; doi: https://doi.org/10.1080/10409230802058320Effective gene therapy with nonintegrating lentiviral vectors; doi: https://doi.org/10.1038/nm1365Exploring the Use of Viral Vectors Pseudotyped with Viral Glycoproteins as Tools to Study Antibody-Mediated Neutralizing Activity; doi: https://doi.org/10.3390/microorganisms13081785


