参考文献:黎海芪. 实用儿童保健学(第2版)[M]. 北京:人民卫生出版社,2022.
❝本文为关于儿童体重与身高生长异常的定义、病因分类及临床评估流程的系统学习记录,仅为个人知识整理。
一、体重生长异常
(一)低体重
1. 定义
体重低于同年龄、同性别儿童体重正常参照值的 <-2SD 或 <P3rd,称为低体重。
2. 病因
- 身材矮小:身高与体重发育平行,如家族性矮小,儿童体重随身高偏低。
- 营养不良:宫内营养不足或严重营养不良的婴幼儿,多伴生长迟缓。
- 慢性疾病:严重心肾疾病、慢性消耗性疾病(如结核、肿瘤、反复呼吸道感染、慢性腹泻等),导致消化吸收功能降低及蛋白质、能量消耗增加。
- 精神因素:不良生存环境、长期精神心理压抑、受虐待等,可影响食欲,导致体重不增或下降;青春期女童可因神经性厌食致体重降低。
(二)体重过重
1. 定义
体重大于同年龄、同性别儿童体重正常参照值均值的 >+2SD 或 >P97th,称为体重过重。
2. 病因
- 营养失衡:摄入能量过多,使身体过多脂肪致体重发育超过身高发育速度。
- 疾病因素:如严重心肾疾病所致水肿;继发性肥胖如库欣综合征、丘脑/垂体和性腺等疾病;某些综合征如Prader-Willi综合征、Laurence-Moon-Biedl综合征和Alstrom综合征等。
二、身高(长)生长异常
(一)矮身材
1. 定义
儿童身高(长)小于同年龄、同性别儿童身高(长)正常均值 -2个标准差(<-2SD) 或 ≤P3rd。
2. 常见病因分类
| |
|---|
| 严重营养不良、慢性感染、先天性心脏病、慢性哮喘、肾脏病、严重地中海贫血、幼年型类风湿关节炎、炎症性肠病、乳糜泻、精神剥夺 |
| |
| 骨软骨发育异常(软骨发育低下、软骨发育不全)、脊柱骨骺发育不良 |
| |
| |
| 21-三体综合征、Turner综合征、Prader-Willi综合征、Williams综合征、Russell-Silver综合征、Noonan综合征 |
| |
3. 各类型矮身材详解
(1)慢性疾病
- 严重营养不良:部分婴幼儿矮小的常见原因,约有2.5%~3%的儿童矮小与此相关。长期喂养不当、慢性疾病及严重畸形所致能量、蛋白质摄入明显不足。儿童矮小水平多为正常临界最低,骨龄可稍落后。营养改善后生长加速,但2~3岁后营养改善,身高常难以完全追赶。
- 继发严重疾病:患严重先天性心脏病、肝病、肾病、慢性腹泻等疾病的婴幼儿生长迟缓。
- 精神心理因素:儿童可因长期精神心理创伤,如父母离异、被遗弃或虐待、遭遇突发事件等,导致生长激素分泌不足,生长迟缓,骨龄落后,第二性征发育延迟。去除不利因素后儿童生长可改善。
(2)特发性矮小(ISS)
- 1975年以前称正常遗传变异矮小,1975年后统一为特发性矮小。
- 儿科临床矮小儿童中约80%为特发性矮小,是儿童期身材矮小的最常见原因,包括家族性或遗传性矮小、体质性发育延迟。
- 临床上往往缺乏低出生体重、出生身长发育史以及疾病史,故多采用“排除法”诊断。ISS可能包括部分未常规检测的遗传异常,如SHOX基因异常。
(3)家族性矮小
- 儿童身高有明显的遗传背景,双亲或双亲之一身高<-2SD。
- 女童 = [母亲身高(cm) + (父亲身高(cm) - 13)] / 2
- 男童 = [父亲身高(cm) + (母亲身高(cm) + 13)] / 2
(4)体质性发育延迟
- 出生时身高与体重正常,生后生长发育速度为正常的低限,骨龄延迟,第二性征发育可延迟,最终身高正常,往往有家族史,男童多见。
(5)继发于宫内发育不良的矮小(SGA)
- 因胎儿生长受限致婴儿出生时体重、身长低于同胎龄儿。
- 约10%的SGA儿童在3岁左右体重、身高仍<P3rd,即SGA持续出生后生长障碍。
- 临床特点为身材匀称性矮小,骨龄正常或略延迟,不伴畸形。
(6)内分泌疾病
- 生长激素缺乏症(GHD):因下丘脑或垂体结构或功能障碍所致生长激素轴失调,可为先天性或获得性缺乏。多数儿童GHD为原发性,可继发于颅脑肿瘤(如颅咽管瘤)或遗传性。原发性GHD多见于男童,智力发育正常,身材匀称,幼稚面容,骨龄<实际年龄2岁,或<-2SD/年龄(正常儿童骨龄±1~2岁),多伴青春期发育延迟。
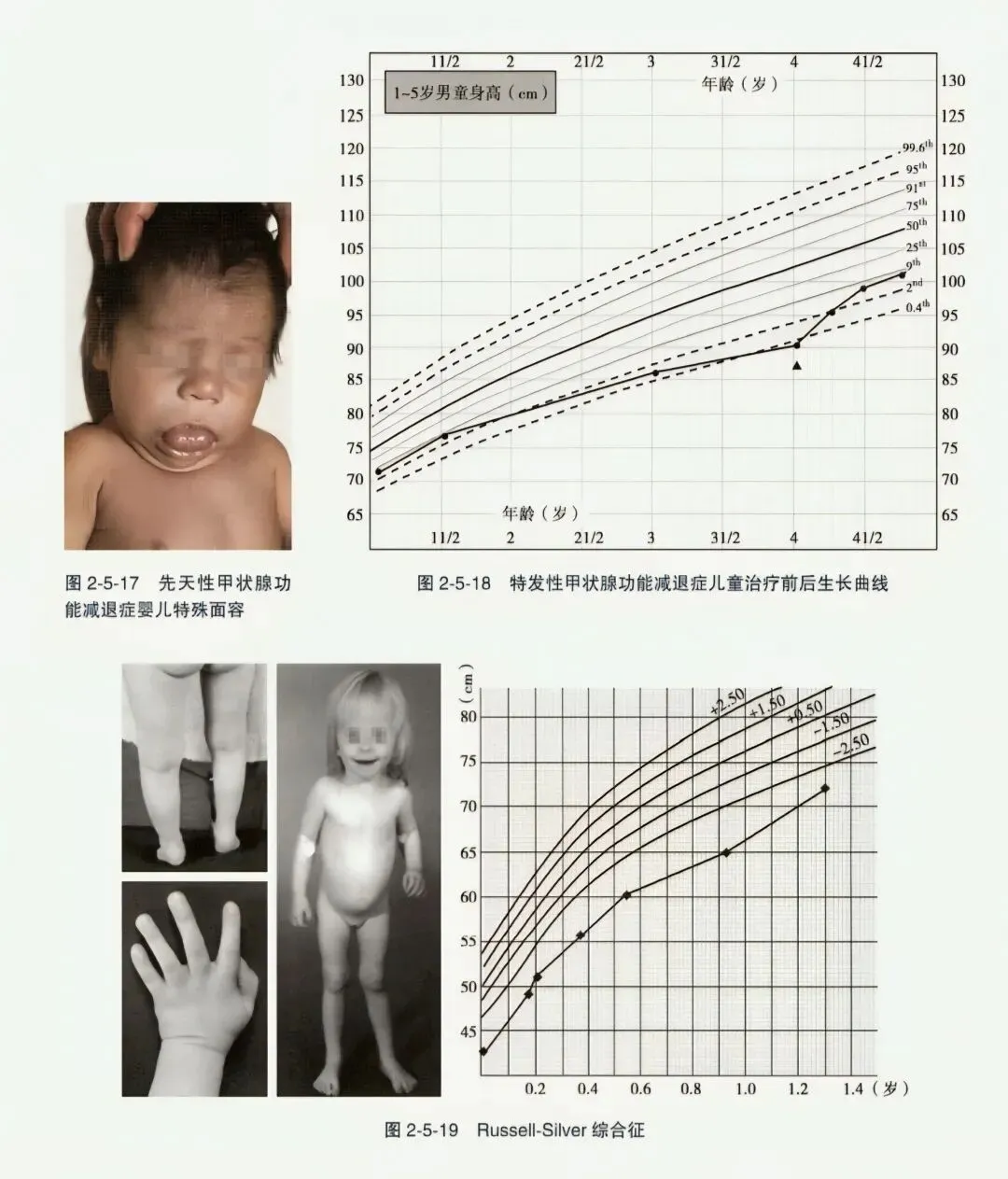
- 先天性甲状腺功能减退症:为甲状腺分泌甲状腺激素不足所致,发病率约为1/2050。严重先天性甲减儿童生长缓慢,为身材比例不匀称矮小(坐高/身高比例幼稚),骨龄显著延迟,黏液性水肿面容(眼距宽、鼻梁宽平、舌大而宽、表情淡漠),皮肤粗糙,智力低下。因甲状腺素(T₄)能从胎盘转运供给患儿,婴儿出生时症状可不典型,如仅表现纳少、黄疸、便秘等,易被忽略。
(7)染色体与基因异常
- Silver-Russell综合征(SRS):低出生体重、特殊面容(倒三角形脸、前额突出、小下颌)、肢体不对称和生长迟缓,近年研究显示与7号染色体为母亲单亲二体有关。
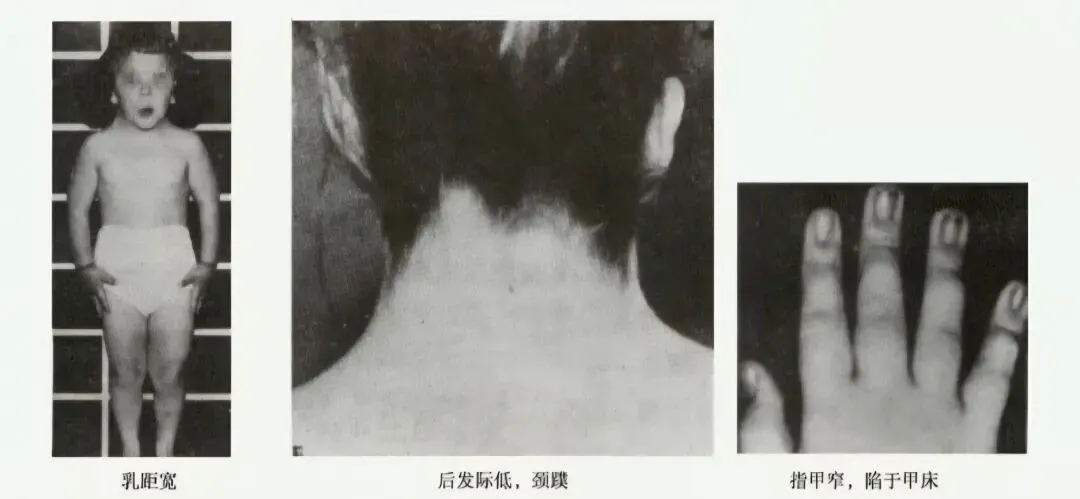
- Turner综合征(TS):又称先天性卵巢发育不全,所有(45,XO)或部分细胞(45,XO/46,XX)缺少一条或部分X染色体,是人类唯一能生存的单体综合征,也是女童矮小最常见的病因之一,发生率为1/5000~1/2000。特征性体征为身材矮小,性发育呈幼稚状态及原发性闭经;颈蹼、肘外翻、发际低、盾状胸、乳头间距增宽、无第二性征;智力多正常。
- 21-三体综合征:又称先天愚型,有共同的特殊面容(眼距宽,小眼裂,双眼外上斜,鼻梁低平,伸舌),生长迟缓,智力发育障碍等多发畸形。
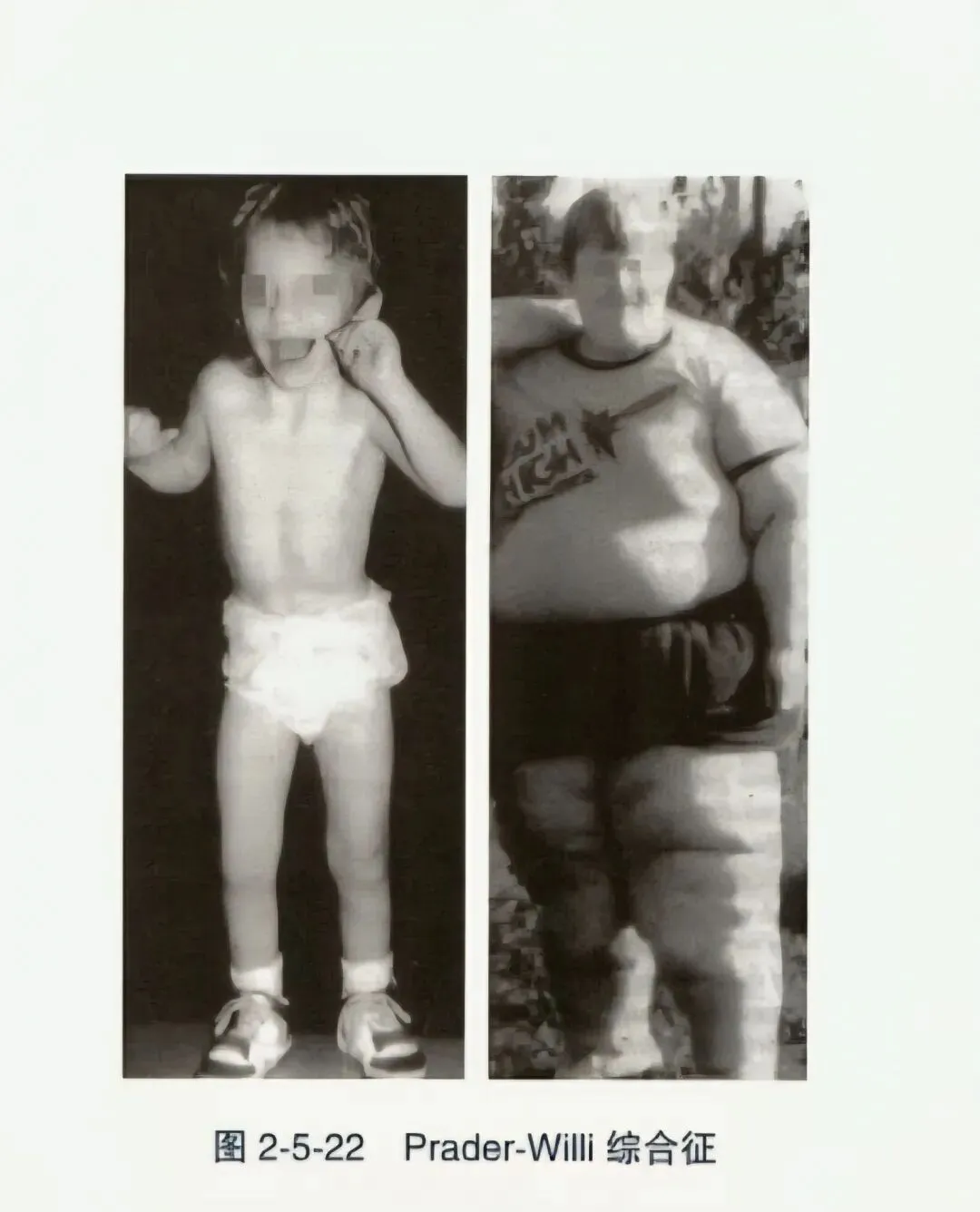
- Prader-Willi综合征(PWS):又称隐睾-侏儒-肥胖-智力低下综合征,发病率为1/25000~1/10000,已证实为母源15号染色体的单亲二体或父源15号染色体上的关键片段发生微小缺失所致。症状与年龄有关,婴幼儿期严重肌无力致喂养困难,1~4岁因食欲亢进而出现中枢性肥胖。特殊面容如狭额、小眼裂、鱼形嘴、小下颌;身材矮小,骨龄正常或延迟,外生殖器发育不良如小阴茎、隐睾、缺乏第二性征。
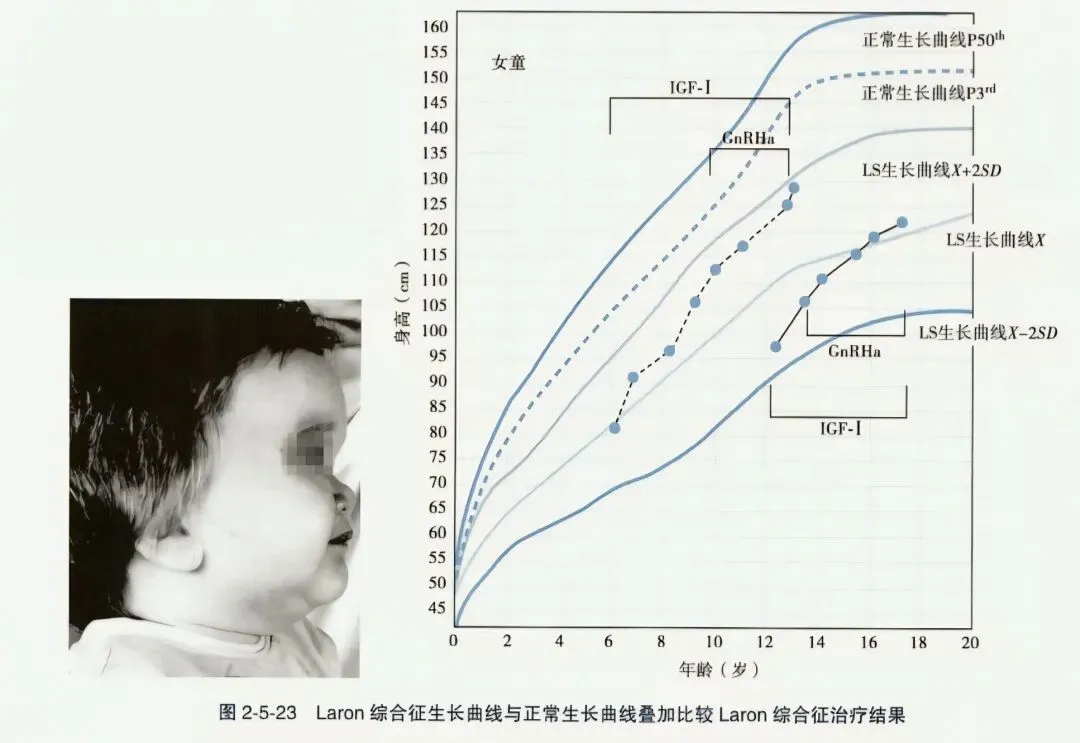
- Laron综合征(LS):又称Laron侏儒、完全性生长激素不敏感、生长激素受体缺乏症,为生长激素受体(GHR)基因突变致胰岛素样生长因子(IGF-1)特别低,系常染色体隐性遗传。临床表现与单纯性生长激素缺乏症相同,严重的生长落后,伴特殊面容(前额突出、大眼睛、塌鼻梁),头发稀软,前囟延迟闭合;血生化特点为高生长激素(GH)、低胰岛素样生长因子-I(IGF-I)和低胰岛素样生长因子结合蛋白-3(IGFBP-3)。
(8)遗传代谢病
- 糖原贮积病(GSD):一类由于先天性酶缺陷所造成的糖原代谢障碍疾病,涉及8种酶,临床表现分12型,I、III、IV、VI、IX型以肝脏病变为主;II、V、VII型以肌肉组织受损为主,发病率约为1/25000~1/20000。I型GSD最常见,为葡萄糖-6-磷酸酶或葡萄糖-6-磷酸酶转运体缺乏。临床表现为肝大、空腹低血糖、乳酸性酸中毒、高血脂、高尿酸血症、骨质破坏、生长迟缓、出血倾向、免疫力低下等。
- 黏多糖贮积症:因溶酶体中某些酶的缺乏不能完全降解黏多糖致黏多糖代谢障碍,临床表现之一是身材矮小。
(9)骨骼发育异常
- 软骨发育不全(ACH):最常见,为长骨骨骺软骨发育不全,表现为四肢短小、躯干相对正常的“短肢型”矮小,头大,智力正常。
- 软骨发育低下(HCH):症状较轻,不典型,易被漏诊或误诊为ISS。
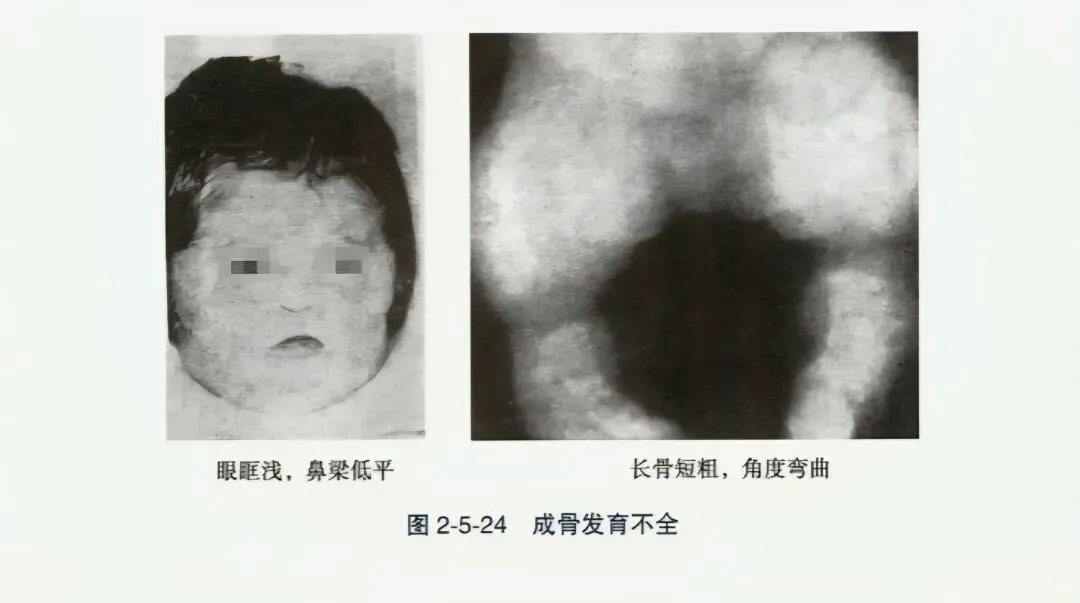
- 成骨不全症(OI):以反复多发性骨折和骨骼畸形为特点,分先天型(重型)和迟发型(轻型)。先天型儿童的骨折始于胎儿或新生儿期,生长迟缓,肢体粗短,伴多种骨骼畸形,常因颅内出血致宫内死亡或早年夭折,骨骼X线多为粗骨型。迟发型成骨不全为婴儿期后一年后出现骨折和骨骼畸形,如脊椎侧弯、后突与胸廓畸形,因脊柱及下肢多发性骨折畸形造成生长迟缓;可伴蓝色巩膜,传导性或神经性耳聋。骨骼X线显示多处陈旧性骨折,细骨型。
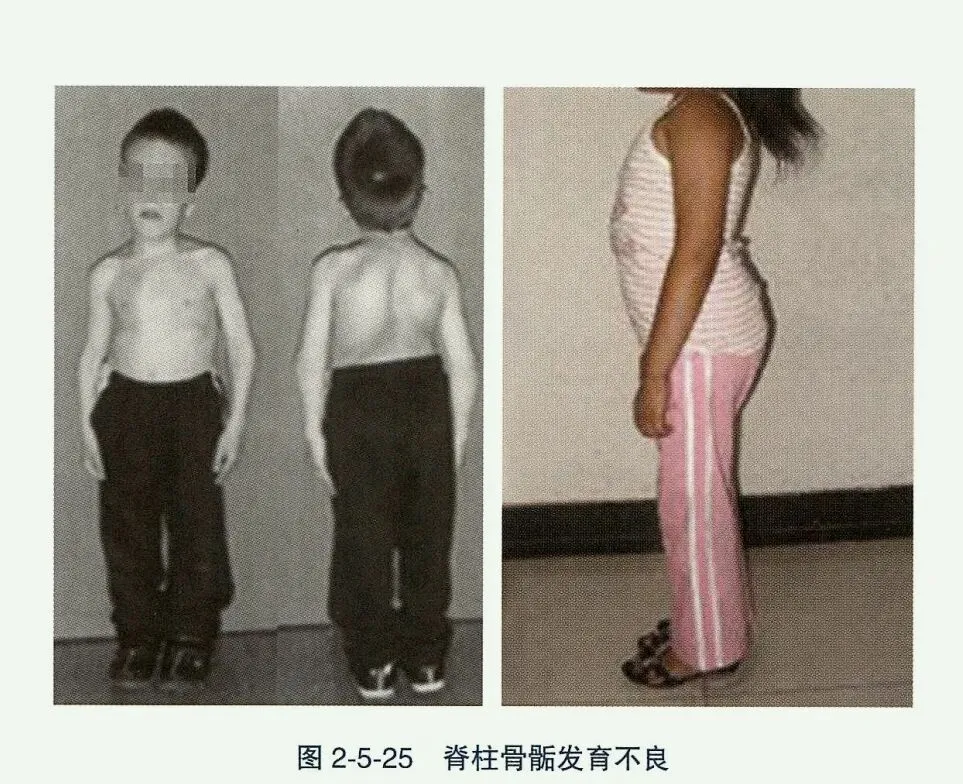
- 脊柱骨骺发育不良(SED):是一组基因突变导致脊柱和骨骺畸形的疾病,主要分先天性脊柱骨骺发育不良(SEDC)和迟发性脊柱骨骺发育不良(SEDT)两大类。主要临床表现包括非匀称性身材矮小(短颈、短躯干)、胸部畸形和早发性关节退行性变,可伴视、听异常。影像学表现为椎体变扁及骨骺发育不良。
(二)超高身材
1. 定义
儿童身高(长)大于同年龄、同性别儿童正常均值加2个标准差(>+2SD)或>P97th为超高身材。与矮小儿童不同,多数超高身材的儿童为正常生长,仅少数为病理性。
2. 常见病因
(1)家族性高身材
- 儿童身高发育与双亲身高一致,如父母身高,子女一般也较高。身高发育主要取决于遗传因素。
(2)性早熟
- 青春期前儿童出现第二性征同时伴有身高提前生长,女童多见。儿童伴有短期生长加速,因性激素提前启动致骨骺闭合提前,最终身高低于遗传靶身高。
(3)染色体异常
- Klinefelter综合征:即先天性睾丸发育不全综合征,常见染色体核型为47,XXY,主要症状为身材高,四肢长,指距>身高5cm,第二性征发育不良,有女性化表现如乳房发育,睾丸小而质硬,97%男性不育,部分智力低下或精神异常。
- 47,XYY综合征:染色体数为47条,性染色体为XYY,故又称YY综合征或超雄综合征。临床症状为身材超高(多大于180cm),智力偏低,脾气暴躁、性情古怪、易激动、固执、具有反社会行为倾向。多数性发育正常,少数性发育不全。
(4)基因异常
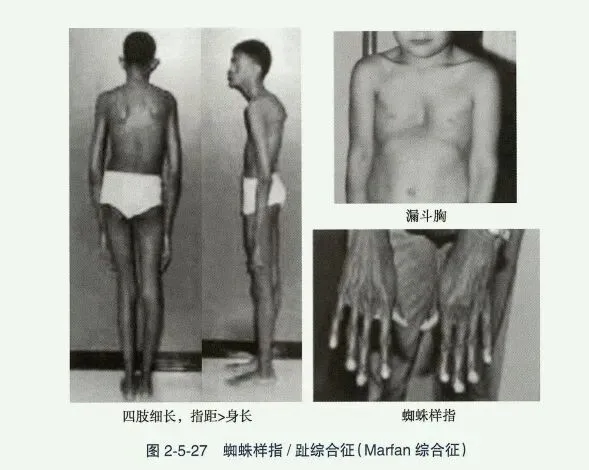
- 马方综合征(Marfan syndrome, MS):又称蜘蛛样指/趾综合征,系常染色体显性遗传性结缔组织疾病,与原纤维蛋白基因(fibrillin-1, FBN1)异常有关。MS是以管状骨细长、蜘蛛样指/趾、眼晶状体移位及先天性心脏病为特征的一组综合征。肢体细长,手和膝过度伸展,智力正常。
- 巨人症:可能与芳基烃交互蛋白质(AIP)基因异常有关,AIP基因异常可引发垂体肿瘤导致生长激素分泌过多。生长激素分泌过多致青少年骨骺闭合延迟形成巨人症,青春期后骨骺已融合则形成肢端肥大症。
三、临床评估流程
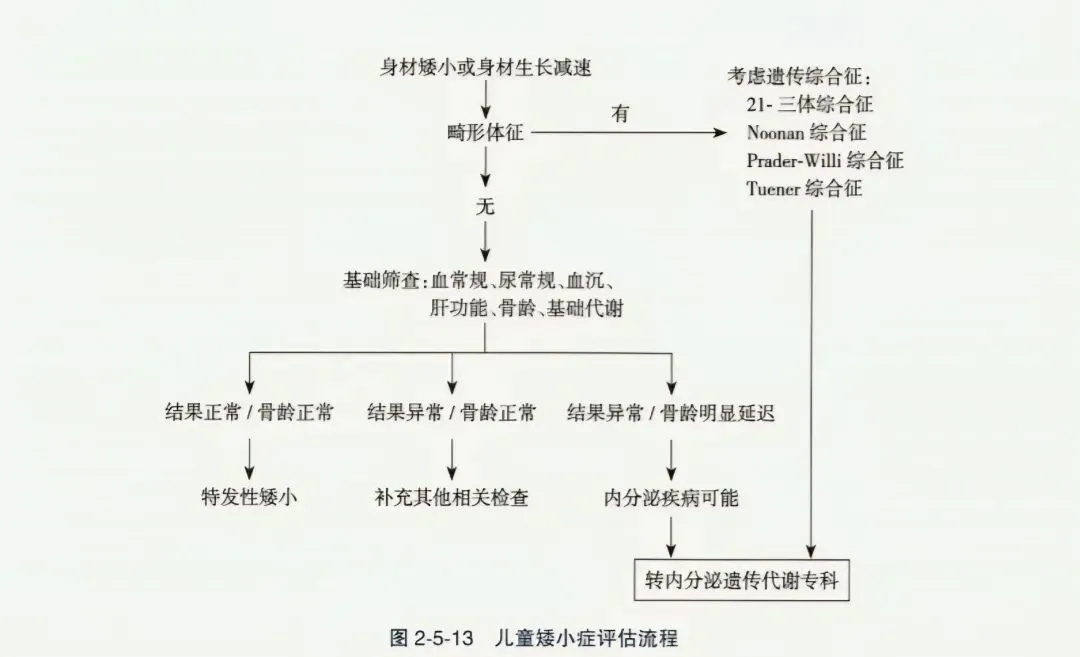
(一)矮身材评估流程
- 首先判断有无畸形体征,如有则考虑遗传综合征(如21-三体、Noonan、Prader-Willi、Turner等)。
- 如无畸形,进行基础筛查:血常规、尿常规、血沉、肝功能、骨龄、基础代谢。
- 结果异常、骨龄明显延迟 → 内分泌疾病可能,转内分泌遗传代谢专科。
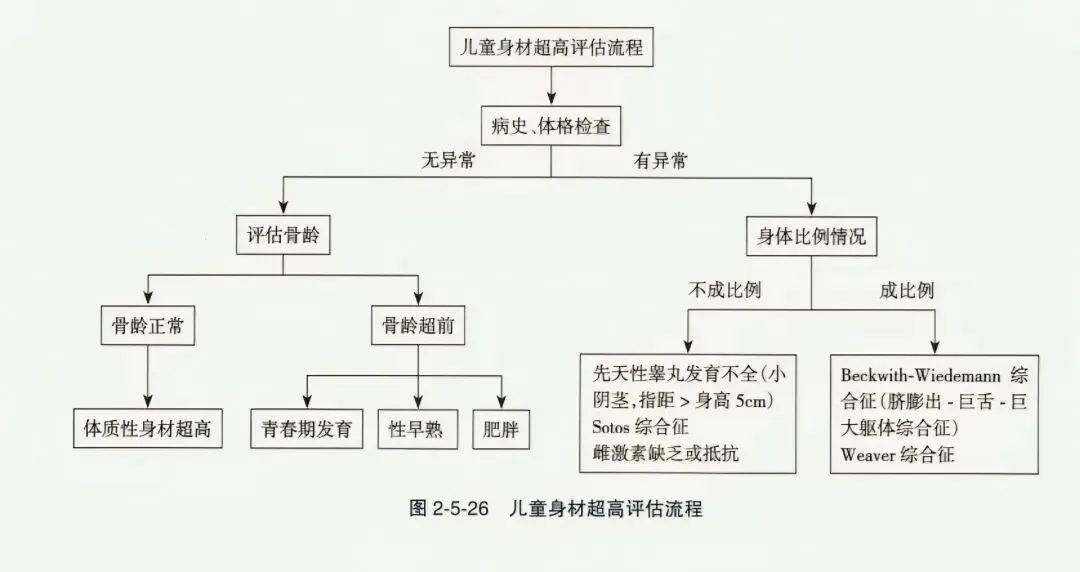
(二)超高身材评估流程
- 不成比例 → 先天性睾丸发育不全(小阴茎,指距>身高5cm)、Sotos综合征、雌激素缺乏或抵抗。
- 成比例 → Beckwith-Wiedemann综合征(脐膨出-巨舌-巨大躯体综合征)、Weaver综合征。
*本文内容整理自相关学习资料,仅为个人学习记录。