学习笔记(25):1145例中国乳腺癌数据揭示新辅助治疗疗效的关键分子标志物
- 2026-05-11 21:46:33

乳腺癌是全球女性中发病率最高的恶性肿瘤之一,新辅助治疗已成为早期及局部晚期乳腺癌的重要标准治疗策略。然而,不同患者、不同分子分型之间的新辅助治疗疗效差异显著,尤其是在中国人群中,缺乏基于大规模真实世界数据的系统性分子证据,始终制约着精准治疗的临床决策。
复旦大学附属肿瘤医院团队2026年4月在Cancer Letters发表研究论文 Integrated Genomic Profiling Identifies Predictive Biomarkers for Neoadjuvant Therapy Response in Chinese Breast Cancer Patients。该研究基于1,145例接受新辅助治疗的中国乳腺癌患者,通过靶向下一代测序技术,系统描绘了新辅助治疗相关的体细胞突变谱,并深入分析了基因组特征与病理完全缓解及治疗耐药之间的关联。
研究不仅揭示了不同分子分型中影响新辅助治疗疗效的关键基因事件,还结合具体治疗方案,探索了基因突变在疗效预测和风险分层中的临床应用价值,为未达到病理完全缓解患者的后续治疗管理提供了新的分子依据。这项工作为中国及亚洲乳腺癌人群的新辅助治疗精准化决策提供了重要的循证支持。


研究方法
本研究系统纳入接受新辅助治疗的中国乳腺癌患者。所有患者在诊断时均依据ER、PR及HER2 状态进行分子分型,并同步收集肿瘤大小、淋巴结状态、分级及增殖水平等关键临床病理信息,为后续分析奠定了坚实基础。
在分子层面,研究对1,145例患者的肿瘤样本进行了高深度靶向基因测序,覆盖近500个与乳腺癌发生发展和治疗反应密切相关的核心基因(484基因和539基因两个套餐版本)。通过严格的质控和人工复核流程,获得了可靠的肿瘤突变信息,并在不同分子亚型中筛选出具有代表性的基因特征,用于评估新辅助治疗疗效和患者预后。
在此基础上,研究引入人工智能方法,在模型构建前,对临床数据进行标准化预处理,包括去除身份识别信息、对分类变量进行编码,并仅纳入信息完整的病例。随后将数据随机划分为训练集(70%)和测试集(30%),用于模型训练与独立验证。特征筛选采用带有L1正则化的逻辑回归模型,并结合十折交叉验证,以识别最稳定、最具预测价值的变量。基于统一建模框架,研究比较了八种机器学习算法,包括提升类模型、树模型、线性模型、神经网络模型及支持向量机等,并通过网格搜索和交叉验证完成参数优化。模型性能以受试者工作特征曲线下面积为核心评价指标,并计算其95%置信区间,同时通过多次自助法重复验证,评估模型预测结果的稳定性与可靠性。

研究设计与队列特征
核心内容:建立一个规模大、分型全、治疗真实的NAT 乳腺癌队列
1. 研究内容:体细胞基因改变、临床病理特征、pCR、DRFS、OS(流程示意图见图1A)
2. 患者临床病理特征(见图1B、表1):
1)分子分型构成:HER2+(36.9%)、HR+/HER2−(39.8%)、TNBC(23.3%);
2)III期患者:63.0%;
3.新辅助治疗方案:
1)HR+/HER2−:90.3%是以化疗为基础、9.7%是以内分泌治疗为基础;
2)HER2+(全部化疗+抗HER2治疗):41.4%是曲妥珠单抗单靶、40.2%是曲妥珠单抗+帕妥珠单抗的双靶、18.5%是曲妥珠单抗+TKI(拉帕替尼/吡咯替尼)
3)TNBC:16.2%是化疗+免疫治疗(卡瑞利珠单抗)、83.8%是单纯化疗
4. pCR结果
1)不同分子亚型pCR率差异显著:HR+/HER2−(7.1%)、HER2阳性(40.2%)、TNBC(28.0%);
5. 随访与生存结局
1) 中位随访时间:56.5个月
2) 总体死亡率:11.6%
3) 中位DRFS:74.3个月(图1C)
4) 复发与死亡时间分布:41.3%的复发发生在治疗后5年内(图1D)、死亡事件主要集中在3–5年;
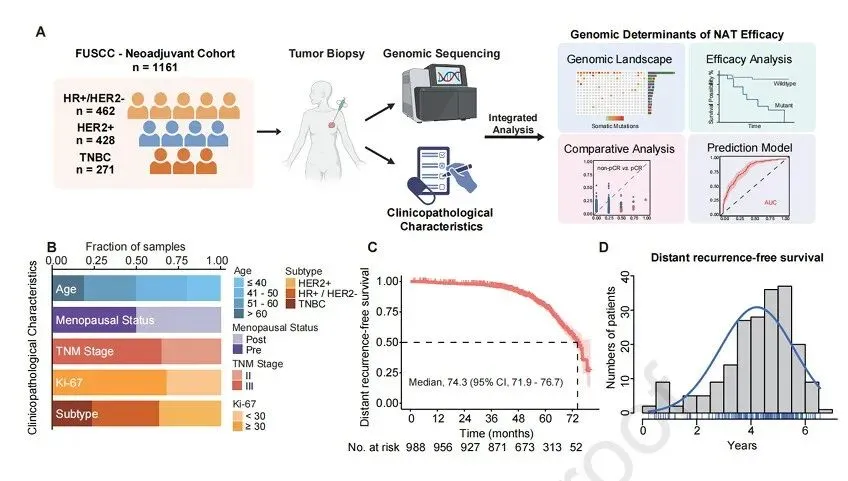
图 1. 研究设计示意图及队列概览。(A) 研究流程示意图。建立一个接受新辅助治疗的乳腺癌队列,并进行靶向测序,通过整合基因组学、临床病理特征及治疗反应分析来鉴定预测性生物标志物。(B) 研究队列的人口学及临床病理特征。展示了手术时年龄、TNM 分期、绝经状态、Ki-67 状态、分子分型以及初诊时的治疗方案。(C) 本研究队列的远处复发无病生存(DRFS)的 KM生存曲线。(D) 本研究队列中远处复发无病生存期的分布情况。

基因组整体分析
核心内容:描绘NAT 队列的突变谱,并识别与 pCR 相关的关键基因与通路
1.高频突变基因(图2A–B)
1)HER2+/TNBC在pCR组中显著富集,而HR+/HER2-主要见于非pCR组
2)Ki‑67指数较高、较早的T分期与pCR显著相关
3)年龄和淋巴结受累情况与疗效无显著关联
4)TMB在pCR组与非pCR组间无显著差异,但特定基因突变和通路改变存在明显不同
5)非pCR组中常见PIK3CA、AKT1等基因突变及PI3K通路改变
6)pCR组则富集TP53、ALK等基因突变,并且AKT1 p.E17K热点突变及p53通路改变更频繁;
1)与TNBC相比,HR+/HER2-显著增加非pCR风险,而HER2+则明显降低非pCR风险
2)Ki‑67指数升高(>30%)与更佳治疗反应显著相关
3)较晚的肿瘤T分期(TⅢ–Ⅳ期相较于TⅠ–Ⅱ期)则预示治疗耐药
4)多因素分析表明PIK3CA、GRIN2A和PIK3R1突变是非pCR的独立不良预测因素;相反,ALK和PGR突变则显著预测获得pCR;
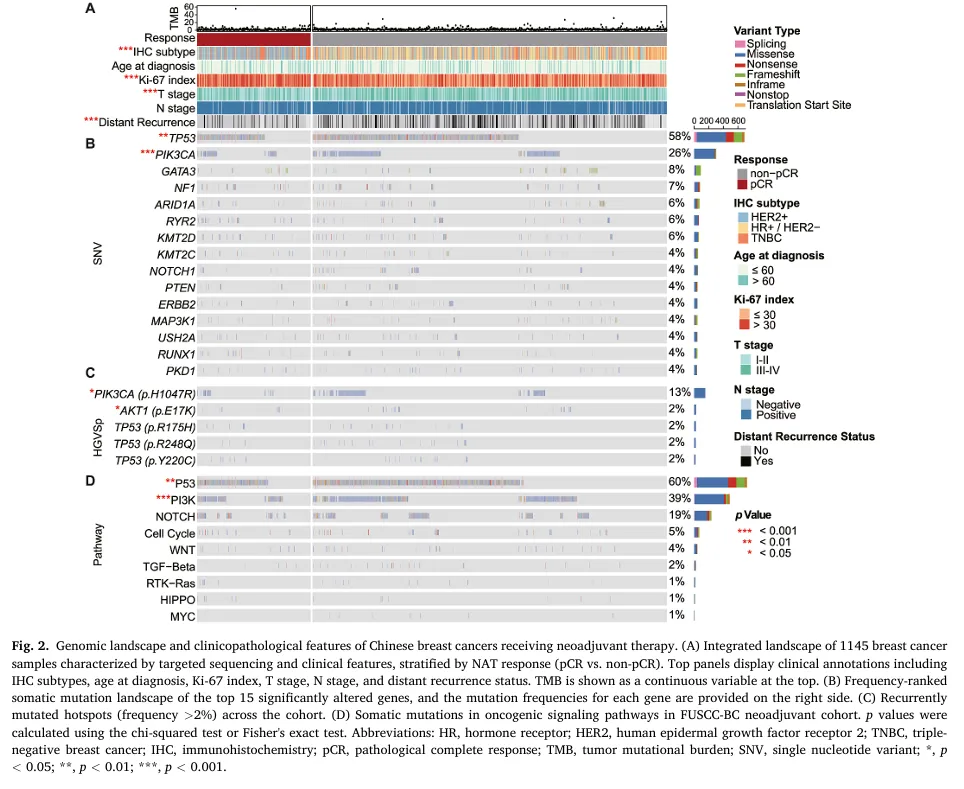
图 2. 接受新辅助治疗的中国乳腺癌的基因组图谱及临床病理学特征。
(A)通过靶向测序和临床特征表征的 1145 例乳腺癌样本 的综合图谱,按照新辅助治疗(NAT)反应(pCR 与非 pCR)进行分层。顶部面板显示临床注释信息,包括 IHC 亚型、诊断时年龄、Ki‑67 指数、T 分期、N 分期以及远处复发状态。TMB 以连续变量的形式显示于最上方。(B)发生频率排序 的前 15 个显著改变基因 的体细胞突变图谱,各基因的突变频率显示于右侧。(C)整个队列中 重复发生的突变热点(频率 >2%)。(D)FUSCC‑BC 新辅助治疗队列 中致癌信号通路的体细胞突变。

分型特异性决定因素
核心内容:不同乳腺癌分型中,决定 NAT 疗效的因素显著不同
1. HR+/HER2−
3. TNBC
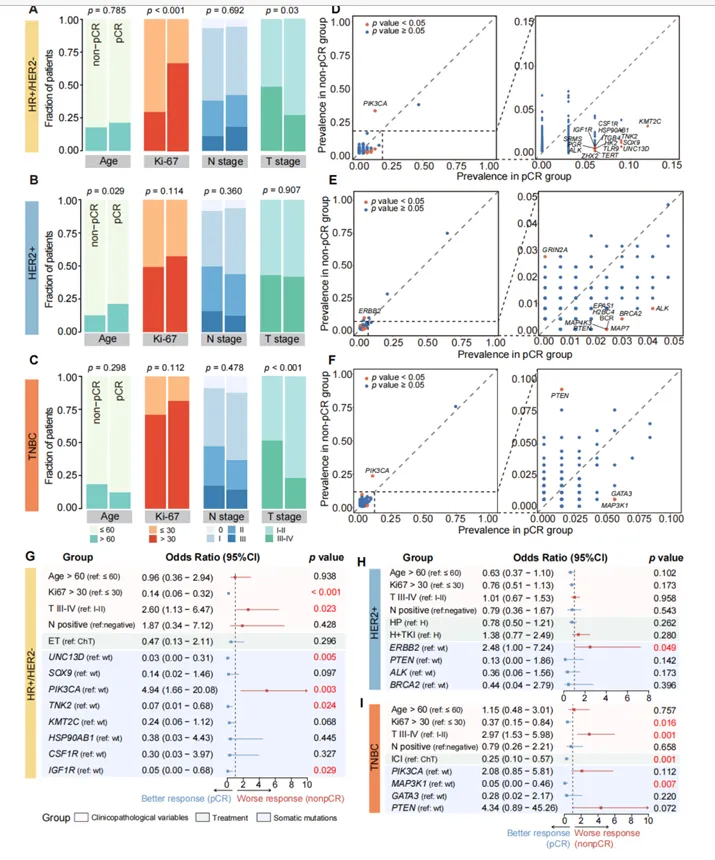
图 3. 乳腺癌中新辅助治疗反应的亚型特异性决定因素。
(A–C)在 HR+/HER2−(A)、HER2+(B)和TNBC(C) 中,比较non-pCR 组与pCR 组的临床特征。(D–F)在 HR+/HER2−(D)、HER2+ (E) 和 TNBC(F) 中,比较non-pCR 组与 pCR 组的基因组突变特征。(G–I)在 HR+/HER2−(G)、HER2+(H) 和 TNBC(I) 中,采用 多变量 Firth 偏倚校正Logistic 回归,评估治疗反应与临床病理特征、治疗方式及基因组特征组合之间的关联。

基因组特征与治疗结局的关联
核心内容:基因标志物的预测作用具有明显“治疗方案依赖性”
1. HER2+(见图 4A)
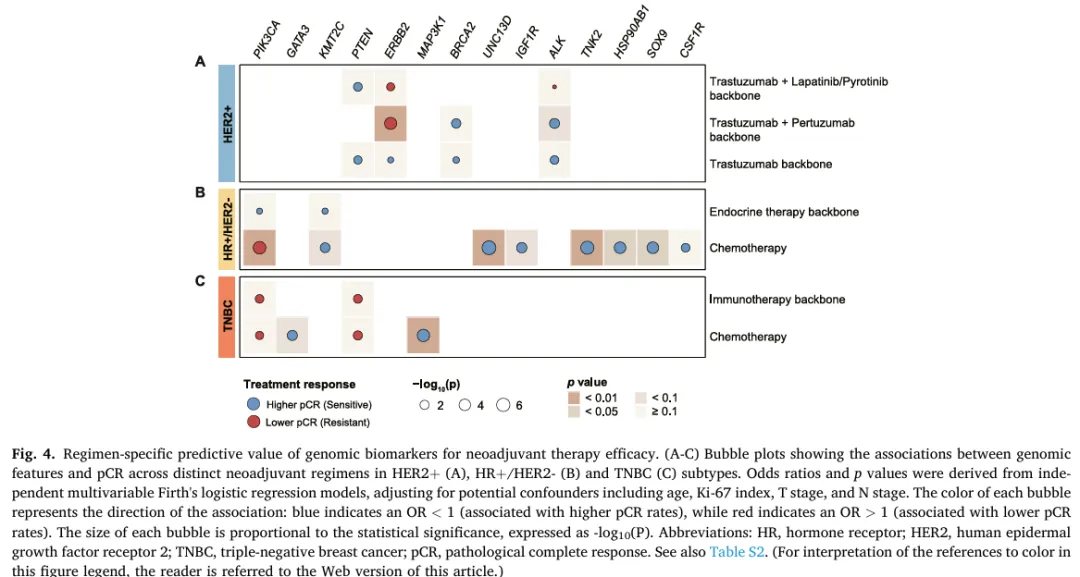
图 4. 基因组生物标志物对新辅助治疗疗效的方案特异性预测价值。
(A–C)气泡图展示了在不同新辅助治疗方案下,HER2+(A)、HR+/HER2−(B) 和 TNBC(C) 亚型中,基因组特征与pCR 之间的关联。比值比(OR)和 p 值来源于相互独立的多变量 Firth Logistic 回归模型,并对潜在混杂因素进行了校正,包括年龄、Ki‑67 指数、T 分期和 N 分期。每个气泡的颜色表示关联方向:蓝色表示 OR < 1(与更高的 pCR 率相关),红色表示 OR > 1(与更低的 pCR 率相关)。每个气泡的大小与统计学显著性成正比,以 −log₁₀(P) 表示。

非pCR组的风险分层
核心内容:在未达 pCR 患者中,基因信息可显著细化复发风险(见图5A)
1.TNBC:DRFS 显著差于HER2+,HR=1.75(见图5A)
2.T 分期晚:T3–T4 相较于 T1–T2:HR=1.55(见图5A)
3.淋巴结阳性:HR=3.00(见图5A)
4. 基因组层面(突变频率大于 2% 的基因被确认是 non‑pCR 队列中的独立预后标志物):TP53(HR=1.74)、PIK3R1 突变(HR=2.49)提示预后不良;STK11 突变(HR=0.12)与较好 DRFS 相关(见图5A)
5 亚型特异风险基因(见图5B):
1)HR+/HER2−:TP53(HR=1.70)、TOP3B(HR=4.00)、BRCA2(HR=2.51)
2)HER2+:TOP2A(HR=4.17)
3)TNBC:SETD2(HR=7.07)、IGF1R(HR=5.42)
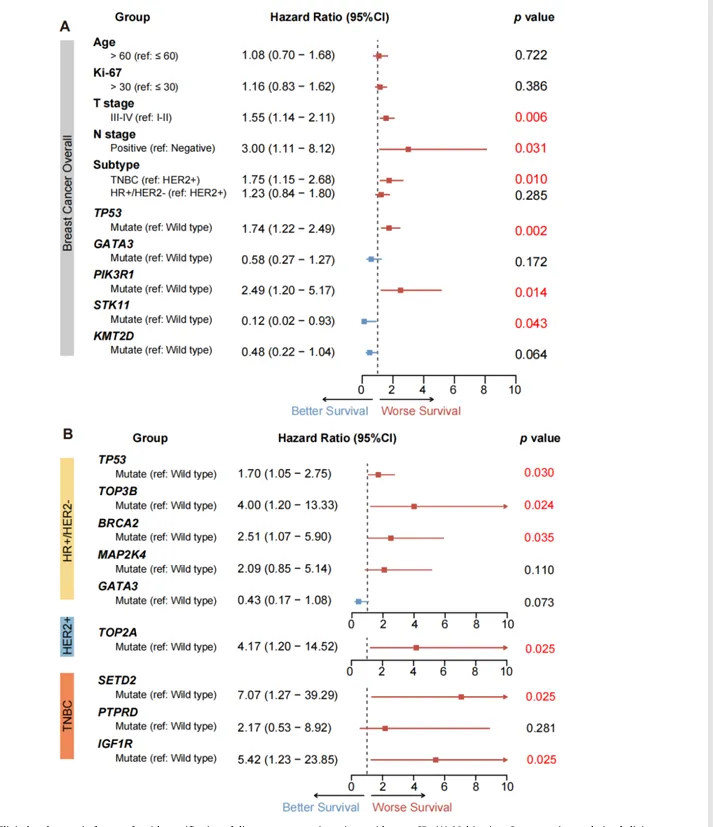
图 5. Non-pCR 患者中用于远处复发风险分层的临床与基因组特征。
(A)在Non-pCR 乳腺癌患者中,采用多变量 Cox 回归分析评估与远处无复发生存期(DRFS)相关的临床病理学和基因组特征。以HR及其 95% 置信区间 进行展示。(B)在 HR+/HER2−、HER2+ 以及 TNBC 亚组中,进行亚型特异性的多变量 Cox 回归分析,评估与 DRFS 相关的基因组驱动因素。在校正潜在混杂因素(包括年龄、Ki‑67 指数、T分期、N分期以及新辅助治疗方案)后,给出HR及其 95% 置信区间。

疗效预测模型
核心内容:整合临床+治疗+基因组构建 NAT 疗效预测模型
1. 整合临床病理特征、治疗方案和基因突变信息,采用 LASSO 筛选特征,并比较 8 种机器学习模型(自适应增强算法AdaBoost、决策树DT、梯度提升决策树GBDT、高斯朴素贝叶斯GNB、逻辑回归LR、多层感知器MLP、支持向量机SVM 以及极端梯度提升算法XGBoost)预测新辅助治疗疗效(见图6A)
2. 多层感知器(MLP)模型性能最佳,在测试集中 AUC 为 0.789,具有较好的预测能力。
3. Ki‑67、T 分期和治疗方案是主要驱动因素,PIK3CA、ERBB2 等基因突变提供了有价值的补充预测信息。
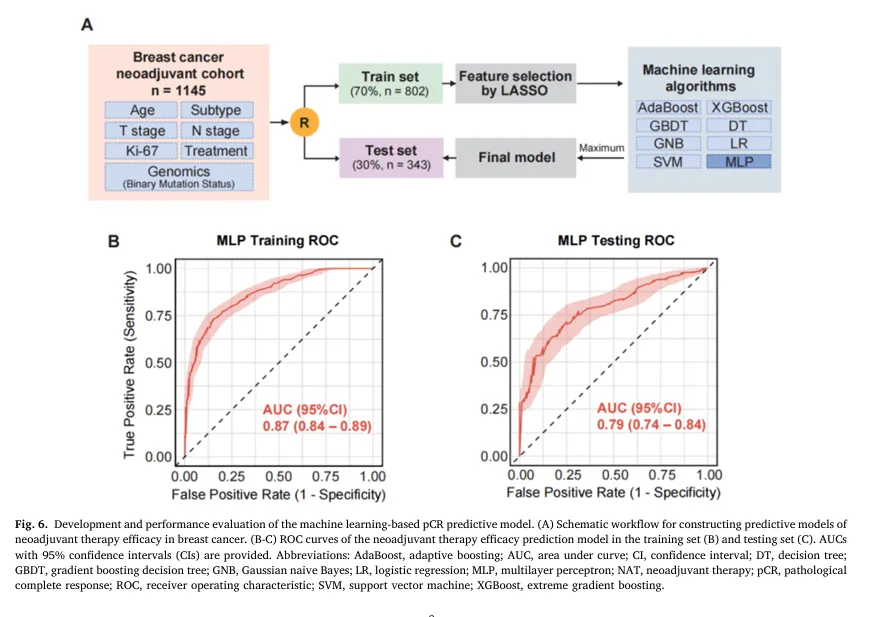
图 6. 基于机器学习的 pCR 预测模型的构建及性能评估。
(A)构建乳腺癌新辅助治疗疗效预测模型的流程示意图。(B–C)新辅助治疗疗效预测模型在训练集(B)和测试集(C)中的 ROC 曲线。同时给出 AUC 值 及其 95% 置信区间(CI)。

本研究系统揭示了中国乳腺癌新辅助治疗人群中显著的人群特异性分子特征及其与疗效的关系。总体而言,中国患者表现出以TP53突变富集、PIK3CA/GATA3/MAP3K1突变频率相对较低为代表的独特突变谱,提示西方人群建立的生物标志物体系并不能简单外推。
在疗效层面,肿瘤对新辅助治疗的应答呈现出明确的分子轴线差异:获得 pCR 的肿瘤更常伴随TP53 通路异常,而non‑pCR肿瘤则显著富集PI3K通路激活相关改变(如 PIK3CA、PIK3R1、PTEN 等)。这种模式在不同分子亚型中具有共性但表现各异,例如HR+/HER2‑肿瘤中PI3K驱动的耐药特征尤为突出,HER2+ 肿瘤中的ERBB2突变影响双靶治疗敏感性,而 TNBC 中PI3K激活与跨方案耐药密切相关,提示PI3K通路是新辅助治疗失败的核心分子基础。
进一步分析表明,在未达到 pCR 的患者中,远处复发风险的决定因素发生了从“解剖学负荷”向“耐药克隆分子特性”的转移。尽管 T、N 分期在整体人群中仍具有预后价值,但在纳入基因突变后,其预测作用在亚型内明显减弱,TP53、BRCA2、TOP2A、SETD2、IGF1R 等突变成为驱动不同亚型non‑pCR患者复发风险的关键因素。这一结果提示残余病灶的生物学本质比残留肿瘤体积更能决定结局。基于此,本研究进一步构建了整合临床指标、治疗方案与基因组特征的预测模型,证明基因突变信息可在传统临床变量之上提供额外、不可替代的预测增量,从而为精准识别耐药人群、优化新辅助及后续强化治疗策略提供了重要依据。

参考资料


