抗体偶联药物(ADC)学习笔记(下):实体瘤渗透、毒副作用、耐药性三大难题及对策
- 2026-04-18 16:57:01
小
寒
学习笔记

本文为个人学习记录,不保证信息准确完整,不构成任何投资建议或行动指南。据此操作,盈亏自负,本人不承担任何责任。
接着上篇《抗体偶联药物(ADC)学习笔记(上):基础概念与五大核心决定因素》今天主要是记录一下ADC 药物三大核心临床困境:实体瘤渗透难、毒副作用、耐药性。
一、ADC实体瘤渗透难题与对策
从生物药理学和药物递送动力学的角度来看,ADC药物在实体瘤中的渗透是一个典型的“反应-扩散-对流”平衡问题。
(一)为什么ADC难以渗透实体瘤?
ADC药物想要到达肿瘤核心细胞,需要跨越血管内皮、穿过细胞外基质,并克服复杂的流体力学阻力。其渗透难主要原因有以下四大屏障:
1. 结合位点屏障效应(Binding-Site Barrier, BSB)
成也高亲和力,败也高亲和力,这是ADC药物特有的“悖论”。 当高亲和力的ADC药物从血管渗出进入肿瘤组织时,会迅速与血管周围(外周)第一层肿瘤细胞表面的抗原结合并发生内吞。 这种极快的“结合与消耗”导致ADC分子在肿瘤外围形成堆积,浓度梯度锐减,极少有游离的ADC能够继续向肿瘤深部无血管区域扩散。
2. 巨大的分子量导致的物理扩散受限
3. 恶劣的肿瘤微环境(TME)流体力学与物理阻力
4. 血管分布的异质性
实体瘤内的血管生长是混乱的,存在大量盲端和无血流灌注的“死胡同”。这意味着肿瘤核心区域往往处于缺氧且完全没有药物递送通道的状态。
(二)解决策略
为了打破上述屏障,目前的药物研发和临床策略主要集中在“改造弹体”、“优化弹头”以及“改造微环境”三个维度:
1. 抗体工程化改造(“瘦身”与“变轨”)
①抗体小型化: 抛弃笨重的完整IgG,开发纳米抗体偶联药物(VDC)、scFv或Fab片段偶联药物。由于分子量降至 15-50 kDa,其组织穿透能力成倍增加。2. 载药与连接子的优化(“隔山打牛”机制)
如果“导弹”实在飞不进深层,那就让“爆炸波”传进去。
①强旁观者效应(Bystander Effect):3. 联合用药改造微环境(“修路”与“松土”)
4. 探索全新偶联平台(XDC)
业界正在将ADC的理念外延,开发多肽偶联药物(PDC)或小分子偶联药物(SMDC)。它们的分子量仅为传统ADC的 1/10 到 1/100,几乎不受微环境物理屏障的限制,能够像常规小分子一样迅速实现实体瘤的深层渗透,同时保留了靶向药物的精准度。

相比于已上市产品,蓝色表示有所提升、黄色表示有所降低
ADC在实体瘤中的渗透是一个复杂的动力学博弈。目前的趋势已经从单纯追求“抗体亲和力越来越高”转向“系统层面的动态平衡”。通过高旁观者效应的载药设计、小型化抗体骨架以及联合微环境调节剂,新一代ADC正在逐步攻克实体瘤深层渗透这一难题。
二、ADC 药物毒副作用
(一)毒性产生途径分析
ADC旨在利用抗体的特异性将高毒性的化疗药物(Payload)精准送达肿瘤细胞,本质上仍是化疗,也会有毒性。其毒性机制非常复杂,通常可以归纳为以下四个核心途径:
1. “找对靶点,但找错地方”:靶向相关毒性 (On-target, Off-tumor Toxicity)
这是最容易理解的一类毒性。虽然 ADC 针对的是肿瘤关联抗原,但绝大多数抗原在正常组织中也有微量表达。肿瘤相关抗原在正常组织低水平表达(HER2 在乳腺上皮、TROP2 在气道上皮、Nectin-4 在泌尿道上皮),当 ADC 进入血液循环后,它会识别并结合表达目标抗原的正常细胞。一旦结合,ADC 就会通过内吞作用进入这些健康细胞,释放毒素并将其杀死。
2. 血液循环中的“走火”与“误食”:脱靶毒性 (Off-target Toxicity)
这是 ADC 最主要、最复杂的毒性来源,主要由 ADC 分子在到达肿瘤之前提前“走火”或被错误摄取引起,引发非特异性细胞杀伤,与抗体的靶向性无关,也是绝大多数 ADC 剂量限制性毒性的核心诱因。
前面也讲到,旁观者效应本是 ADC 的核心抗肿瘤优势:肿瘤细胞内释放的载荷可穿透细胞膜,杀伤周围不表达靶抗原的异质性肿瘤细胞。但当载荷从靶抗原阳性的正常细胞中释放后,会扩散至周围正常组织细胞,引发级联式的正常组织损伤,显著放大毒性。 (毒素的脂溶性越强,穿透细胞膜的能力越强,旁观者效应越明显,潜在的组织毒性也可能越高。)
例如,肺泡上皮细胞低表达 HER2,ADC 结合后释放的 DXd 载荷(DS-8201 的载荷)可扩散至周围肺泡上皮、血管内皮细胞,加重肺部炎症与纤维化,是 ILD 发生进展的核心机制之一。
3. 免疫原性介导的毒性
ADC 是人工合成的大分子生物制剂,其抗体的人源化程度、连接子 - 载荷复合物的异源性,均可诱导机体产生抗药物抗体(ADA),进而引发多维度的免疫相关毒性。就是有的人会对花粉、海鲜过敏一样,把ADC这个药当成外来的“细菌”,让身体免疫系统攻击他。这就会带来两方面问题:
一是免疫复合物沉积与超敏反应:ADA 与 ADC 结合形成免疫复合物,随血液循环沉积在肾小球基底膜、血管壁、关节滑膜等部位,激活补体系统,引发 III 型超敏反应,导致肾小球肾炎、血管炎、关节炎等;同时可介导 I 型超敏反应,引发输液相关反应,轻者出现发热、皮疹、寒战,重者出现过敏性休克。 二是毒性放大效应:ADA 结合 ADC 后,会加速单核 - 巨噬细胞系统对 ADC 的吞噬清除,在降低药物疗效的同时,导致大量 ADC 在肝脏、脾脏的巨噬细胞中富集,载荷集中释放,显著加重肝、脾等器官的毒性损伤。
4、其他特殊毒性途径
一是载荷的非分裂细胞毒性。多数ADC 载荷针对快速分裂的肿瘤细胞设计,但部分载荷存在非周期依赖性毒性,可损伤静息期的正常细胞。
二是肿瘤裂解综合征。ADC 对肿瘤细胞的高效杀伤,可导致大量肿瘤细胞短时间内裂解,释放细胞内的钾离子、尿酸、磷酸盐等物质进入血液循环,引发高钾血症、高尿酸血症、急性肾功能衰竭、心律失常,严重时可致死,多见于肿瘤负荷高、对 ADC 高度敏感的患者。
三是肠道菌群相关的毒性放大。部分 ADC 载荷经胆汁排泄进入肠道后,可破坏肠道菌群平衡,导致肠道屏障功能受损、内毒素入血,引发全身炎症反应;同时,肠道菌群的酶类可进一步代谢载荷,生成毒性更强的代谢产物,加重胃肠道毒性与全身毒性。
目前 ADC 的研发方向正在从单纯的“强效”转向“精准平衡”。例如,开发可裂解但更稳定的连接子,或者筛选亲和力适中(减少对正常组织低表达抗原的捕获)的抗体,都是为了在杀伤肿瘤的同时,让这枚“导弹”不要误伤平民。
(二)解决策略
1. 针对“靶向相关毒性 ”的解决策略
既然“找对靶点但找错地方”,药企的思路是让 ADC 只有在肿瘤微环境(TME)中才被激活,或者提高靶向的门槛。
策略 1:前体抗体 / 掩蔽抗体技术 (Probody/Masked Antibody)
机制: 用一段多肽把抗体的结合区“遮挡”起来,使其在正常血液循环中不结合正常组织。只有到达肿瘤微环境,被肿瘤特异性高表达的蛋白酶(如 MMPs、uPA)剪切掉遮挡物后,ADC 才恢复结合能力。
代表药物:CX-2029 (CytomX 开发的靶向 CD71 的 Probody ADC)。CD71 在正常细胞广泛表达,直接成药毒性极大,采用掩蔽技术后大幅拓宽了治疗窗口。
策略 2:双特异性 ADC (Bispecific ADC)
机制: 要求 ADC 必须同时结合肿瘤细胞表面的两个不同抗原(或同一抗原的两个不同表位)才能高效内吞。正常细胞极少同时高表达这两种抗原,从而实现精准打击。
代表药物:BL-B01D1 (百利天恒,EGFR x HER3 双抗 ADC);Zanidatamab zovodotin (ZW49) (Zymeworks,HER2 双表位 ADC)。
2. 针对“脱靶毒性”的解决策略
这是目前药企优化的重中之重,核心在于“提高稳定性”和“隐身”。
策略 1:开发极度稳定的连接子与定点偶联技术
机制: 摒弃早期随机偶联(容易产生不稳定的大分子聚合物),采用定点偶联技术(如引入非天然氨基酸、半胱氨酸突变等),使得连接子在血液中坚如磐石,只有在溶酶体特定酶的作用下才裂解。
代表药物:ARX788 (新码生物/Ambrx,HER2 ADC)。采用非天然氨基酸定点偶联(pAF),其连接子在血液中极其稳定,临床数据中几乎未观察到脱发、严重胃肠道毒性等传统 ADC 常见的系统性脱靶毒性。
策略 2:Fc 段沉默 / 改造工程 (Fc Silencing)
机制: 针对单核-巨噬细胞系统的错误吞噬,药企通过基因工程改造抗体的 Fc 段,消除或极大降低其与巨噬细胞表面 FcγR 的结合力,防止 ADC 在肝、肺巨噬细胞中蓄积。
代表药物: 目前许多处于临床前或早期临床的新一代 ADC(如靶向免疫细胞的 ADC 或非肿瘤 ADC)都开始常规引入 Fc 沉默突变(如 LALA 突变)。
策略 3:优化 DAR(药物抗体比)
机制: 传统观念认为毒素挂得越多越好(高 DAR),但高 DAR 会导致 ADC 疏水性增加,更容易在循环中被非特异性清除或引发免疫反应。如今倾向于均一且适中的 DAR。
代表药物:Datopotamab deruxtecan (Dato-DXd)。相比 DS-8201 (DAR=8),Dato-DXd 将 DAR 降低至 4,在保证疗效的同时,显著降低了间质性肺病 (ILD) 和血液毒性的发生率。
策略 4:优载荷回收型 PR‑ADC 平台(荣昌生物全球首创)
机制:针对传统 ADC循环中游离载荷泄漏这一脱靶毒性核心源头,在抗体骨架上融合游离毒素特异性捕获模块,可精准识别并结合血液中泄漏的游离毒性分子,形成复合物后被机体快速清除,从源头降低全身游离毒素暴露;同时不影响肿瘤细胞内的正常载荷释放与杀伤功能,实现 “靶向杀伤 + 毒素回收” 双重作用,显著降低神经毒性、骨髓毒性等系统性脱靶不良反应,大幅拓宽治疗窗口。
代表药物:暂无。另外目前公开信息,荣昌生物该平台,只针对微管蛋白抑制剂MMAE,如果未来可以扩展到DNA 损伤剂,那该平台价值将会有巨大提升。
3. 针对“免疫原性介导毒性”的解决策略
策略:全人源化抗体与高同质性
机制: 使用全人源化抗体替代嵌合抗体;通过定点偶联技术确保批次间的极高同质性,减少由于药物聚合物引发的机体免疫识别。
代表药物: 几乎所有新一代处于临床阶段的 ADC(如 SKB264 (芦康沙妥珠单抗))均采用了高度人源化的抗体骨架和优化的均一性工艺。
4.针对“特殊毒性(载荷毒性)”的解决策略
从“剧毒化学品”转向“温和且多机制”的新型载荷。
策略 1:拓扑异构酶 I 抑制剂的全面崛起
机制: 早期 ADC 爱用微管蛋白抑制剂(MMAE/MMAF)和 DNA 损伤剂(PBD),毒性极强。现在的趋势是使用毒性相对较弱、具有中等细胞穿透能力的拓扑异构酶 I 抑制剂(如 DXd、SN-38 等),靠增加肿瘤内药物浓度来杀癌,减少了对外周神经和静息细胞的伤害。
代表药物: 第一三共的 DXd 平台(DS-8201, DS-1062, HER3-DXd 等);科伦博泰的毒素-连接子平台(SKB264,采用具有一定旁观者效应但不至于过度毒性的由贝洛替康衍生的毒素)。
策略 2:抗体-降解剂偶联物 (DAC) 与免疫刺激偶联物 (ISAC)
机制: 彻底更换载荷类型。DAC 挂载的是 PROTAC(蛋白降解剂),只降解特定致病蛋白而不直接毒死细胞;ISAC 挂载的是免疫激动剂(如 TLR 激动剂),只在肿瘤局部激活免疫系统,而非直接化疗毒杀。
代表药物:ORM-5029 (Orum Therapeutics 开发的 HER2-GSPT1 降解剂 ADC);SBT6050 (Silverback 开发的 HER2-TLR8 ISAC)。
将上述应对策略整理如下表:
| 毒性来源 | 核心难点 | 药企破局策略 (技术路线) | 代表药物/平台 |
| 靶向正常组织毒性 | 靶点在正常组织有表达 | 掩蔽抗体技术 (Probody)、双抗 ADC (提升内吞门槛) | CX-2029 (Probody), BL-B01D1 (双抗ADC) |
| 脱靶系统性毒性 | 连接子断裂、巨噬细胞吞噬 | 定点偶联技术、高稳定酶促裂解连接子、Fc段沉默 | ARX788 (定点偶联), Dato-DXd (DAR 4 优化) |
| 免疫原性毒性 | ADA抗药抗体产生 | 全人源抗体骨架、提升产品均一性以减少聚合物 | 新一代ADC通用标准 |
| 载荷特异性毒性 | 神经毒性、VOD、重度骨髓抑制 | 迭代毒素(转用 Topo I 抑制剂)、开发 DAC (降解剂)及 ISAC | DS-8201 (DXd载荷), ORM-5029 (DAC) |
知识扩展:ADC 的临床毒性并非由单一组分决定,而是由靶点、连接子与载荷共同构成的“生化系统”与人体组织相互作用的综合结果。 以 TROP2-ADC 为例,尽管 SG 与 Dato-DXd 靶向一致且载荷类别相同,但两者展现了截然不同的毒性谱系:SG 凭借 高 DAR 值(平均约 7.6)与易外排的 SN-38 载荷,表现出类似传统化疗的骨髓抑制与肠道损伤;而 Dato-DXd 通过 降低 DAR 值(4.0) 虽然改善了血液安全性,却因 DXd 载荷 特有的细胞膜通透性及分布特性,诱发了口腔炎、干眼症以及极具器官特异性且高风险的间质性肺病(ILD)。这充分证明,连接子的稳定性、DAR 值的平衡以及载荷微观结构的差异,能够彻底重塑药物在体内的药代动力学(PK)分布与非特异性内吞偏好,使得即便“同靶同类”的导弹,也会因其内部精密构造的微调而导致完全不同的“误伤”后果。
ADC 药物的研发已经度过了“唯毒性论”的蛮荒时代,进入了微雕时代的“治疗窗口之战”。未来的 ADC 会越来越像一枚拥有“敌我识别系统(掩蔽技术)”、“保险栓(稳定连接子)”和“精确爆炸当量(优化 DAR 与新型载荷)”的智能导弹。
三、ADC药物耐药性
(一)耐药性生物药理学分析
ADC的耐药性是一个复杂的多因素、多步骤过程。由于ADC发挥药效需要经历“循环、结合、内化、释放、杀伤”等多个独立环节,肿瘤细胞可以在其中的任何一个节点进化出逃逸机制。以下是按照ADC药物在体内的药理学作用轨迹,对耐药性机制的拆解:
1. 抗原识别与内化层面(第一道防线)
这是ADC发挥靶向作用的关键前提。如果药物无法有效进入细胞,后续的毒素释放就无从谈起。
①靶抗原下调或丢失(Antigen Downregulation/Loss):2. 溶酶体与连接子切割层面(释放障碍)
ADC进入细胞后,必须依赖溶酶体的酸性环境和酶类将连接子(Linker)切断,释放毒素。
①溶酶体pH值升高(Lysosomal Alkalinization):3. 毒性载荷(Payload)作用层面(终末杀伤失效)
即使Payload成功释放到细胞质中,细胞依然可以通过传统的化疗耐药机制进行反击。
①药物外排泵过度表达(Efflux Pump Upregulation):4. 肿瘤微环境(TME)与系统性因素
除了细胞内部机制,生物体层面的障碍也会导致“药代动力学(PK)/药效学(PD)”层面的耐药。
①物理屏障(Stromal Barriers):(二)应对策略探讨
1. 针对“抗原丢失与内化受阻”的对策
当肿瘤细胞通过减少“入口”来逃避打击时,药企的策略是增加“备选入口”或提高“抓取效率”。
策略 1:双特异性 ADC (Bispecific ADC)
办法:双表位型 (Dual-Epitope):同时靶向同一抗原的两个不同位点。逻辑: 提高抗原结合的稳定性和内吞速率,同时减少受体脱落(Shedding)带来的影响。
双抗原型 (Dual-Antigen):同时靶向两种不同的抗原(例如 EGFR 和 c-MET,或 HER2 和 HER3)。逻辑: 覆盖更广的肿瘤细胞群,防止单一抗原丢失导致的耐药。
抗体-毒素-干扰型:一种抗原负责导航,另一种抗原负责抑制肿瘤生长信号。逻辑: 除了毒素杀伤外,通过阻断双通路信号实现协同抑瘤。
代表药物:Merus的MCLA-129 (靶向 EGFR 和 c-MET);Zymeworks的ZW49 (靶向 HER2 的两个不同表位);百利天恒BL-B01D1(靶向EGFR x HER3)。
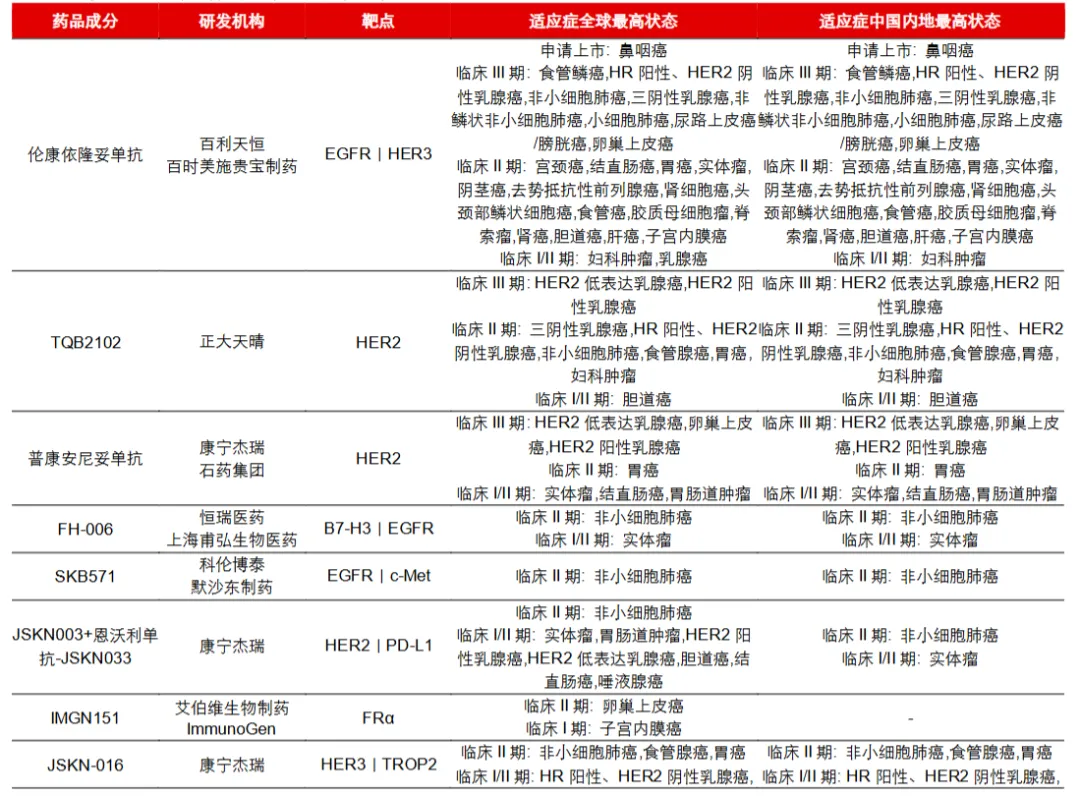
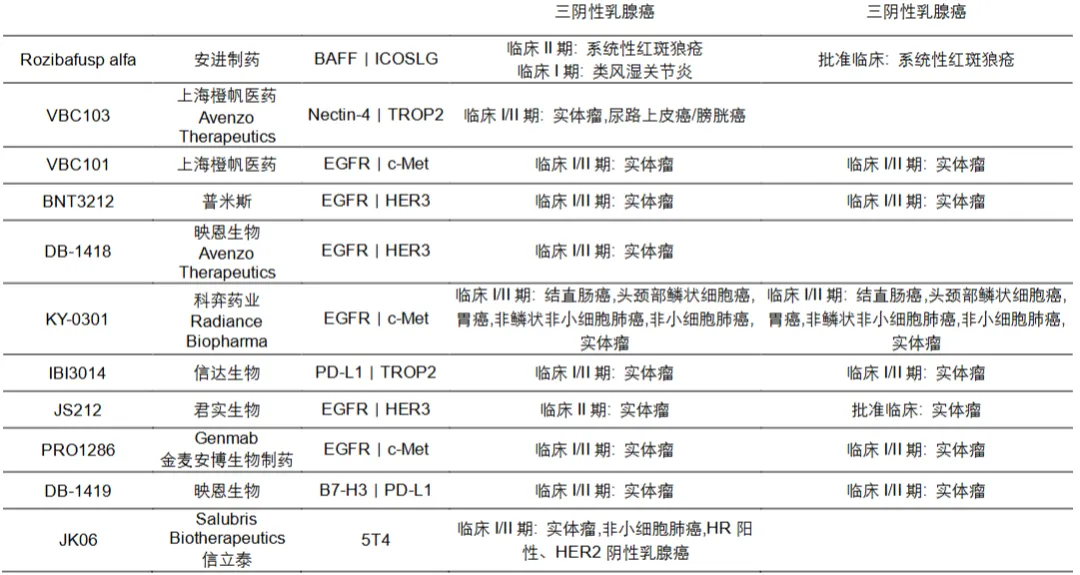
数据截止到2025年年底
策略 2:利用“非内化型”ADC
办法: 研发不依赖内吞也能释放毒素的 ADC。通过在肿瘤微环境(TME)中特异性裂解连接子,在细胞外直接释放具有强旁观者效应的毒素。
代表药物: 这种策略目前多见于靶向肿瘤基质(如 FAP、Tenascin-C)的研发管线中。
策略 3:联用上调靶点表达的药物
办法: 配合小分子药物(如组蛋白去乙酰化酶抑制剂 HDACi)来诱导抗原重新表达。
2. 针对“溶酶体释放障碍”的对策
当溶酶体变得“不给力”时,药企尝试跳过溶酶体,在其他地方“爆破”。
策略:开发“溶酶体外”可裂解连接子
办法: 研发对谷胱甘肽 (GSH) 敏感(二硫键)或对活性氧 (ROS) 敏感的连接子。这些物质在肿瘤细胞质中浓度极高,ADC 进入细胞质后即可裂解,不依赖溶酶体的酶或酸性环境。
代表药物: 许多基于二硫键的 ADC 正在测试中,以应对溶酶体酶下调导致的耐药。
3. 针对“毒素外排(P-gp/MDR1)”的对策
这是目前最成熟的耐药破解方向。
策略 1:疏水性改进与亲水性连接子
办法: 许多毒素(如 MMAE)因为是疏水性的,很容易被外排泵抓住。通过引入PEG(聚乙二醇)或亲水性多肽连接子,可以降低 ADC 的疏水性,使代谢后的毒素更难被 P-gp 识别和排出。
代表药物:Trodelvy (SG) 的连接子设计就考虑了降低疏水性。
策略 2:使用不是 P-gp 底物的新型载荷
办法: 筛选天然不被 P-gp 泵出的毒素分子。
代表药物:Enhertu (DS-8201)。其载荷 DXd 的设计精妙之处在于:它是 P-gp 的弱底物,极难被泵出。这就是为什么 DS-8201 在 T-DM1 耐药(通常由外排泵引起)的患者中依然高效的核心原因。
4. 针对“Payload 靶点突变与凋亡逃逸”的对策
策略 1:载荷切换(Payload Switching)
办法: 当微管蛋白抑制剂(MMAE/DM1)耐药后,改用 DNA 损伤剂(DXd/SN-38)。通过更换“弹头”类型,彻底绕过原有的耐药机制。
代表药物: 临床上常在 T-DM1(微管类)进展后,序贯使用 DS-8201(Topo I 类)。
策略 2:双载荷 ADC:
办法:两种机制毒素协同,阻断单靶点逃逸。
代表药物:康弘药业 KH815,载荷组合:拓扑异构酶 I 抑制剂(TOP1i)+ RNA 聚合酶 II 抑制剂,DNA/RNA 双通路,逆转 P-gp 耐药。
5. 针对“物理屏障与微环境”的对策
策略 1:小型化 ADC 架构
办法: 放弃巨大的全抗体骨架(150kDa),改用纳米抗体(VHH)、小分子偶联药物(SMDC)或多肽偶联药物(PDC)。分子量小,渗透力强,能钻进致密的肿瘤组织。
代表药物:Pepaxto (melflufen)(虽然因多种原因退市,但代表了 PDC 的方向);Bicycle Toxin Conjugates (BTC)。
策略 2:联合抗血管生成药物
办法: 联用贝伐珠单抗等药物使肿瘤血管“正常化”,降低组织间压,增加 ADC 的渗透。
ADC 耐药性对策速查表
| 耐药环节 | 药企解决办法 | 关键药物/技术 |
| 抗原丢失/降低 | 双靶向 ADC、联合治疗诱导抗原表达 | BL-B01D1 (EGFR/HER3), ZW49 |
| 内化效率极低 | 非内化连接子 (TME 裂解) | 研发中 (靶向 Stroma) |
| 溶酶体功能障碍 | 胞质敏感连接子 (GSH/ROS 触发) | 各种二硫键连接子 ADC |
| 外排泵 (MDR1) | 降低疏水性、使用 P-gp 弱底物载荷 | DS-8201 (DXd), Trodelvy |
| Payload 机制耐药 | 更换载荷类别,双载荷 | SKB264,康弘药业 KH815(DNA+RNA 双通路杀伤) |
| 渗透性差 | 小型化骨架 (PDC / 纳米抗体) | Bicycle Toxin Conjugates |
知识扩展:《ADC诱导多倍体巨癌细胞形成的机制及其在耐药中的作用》
四、 总结与展望:从“拼图时代”走向“系统微雕”
总体来看,ADC药物的研发已经彻底告别了早期的“拼图时代”(单纯追求高亲和力抗体与极毒载荷的简单拼接),全面迈入了“系统微雕”与“动态博弈”的新纪元。实体瘤渗透难、毒副作用复杂、以及不可避免的耐药性,本质上是ADC药物在体内复杂药理学与动力学轨迹下的一场连锁反应。 这三大困境并非孤立存在,它们的解决策略也常常互为表里:①为了克服渗透难,我们引入了强旁观者效应的载荷;②但强旁观者效应与不稳定连接子的叠加,又放大了系统性毒性;③而为了控制毒性下调载荷毒性或降低靶点亲和力,又可能给肿瘤细胞留下耐药逃逸的喘息之机。
因此,新一代的ADC研发正是在这三者构成的“不可能三角”中寻找完美的动态平衡。从单靶点向双靶点/多特异性迈进(解决抗原丢失),从传统化疗毒素向双载荷或蛋白降解剂(DAC)演变(解决外排耐药与特殊毒性),再到不断引入前体掩蔽技术与微环境调控策略(提升肿瘤特异性渗透与安全性)。
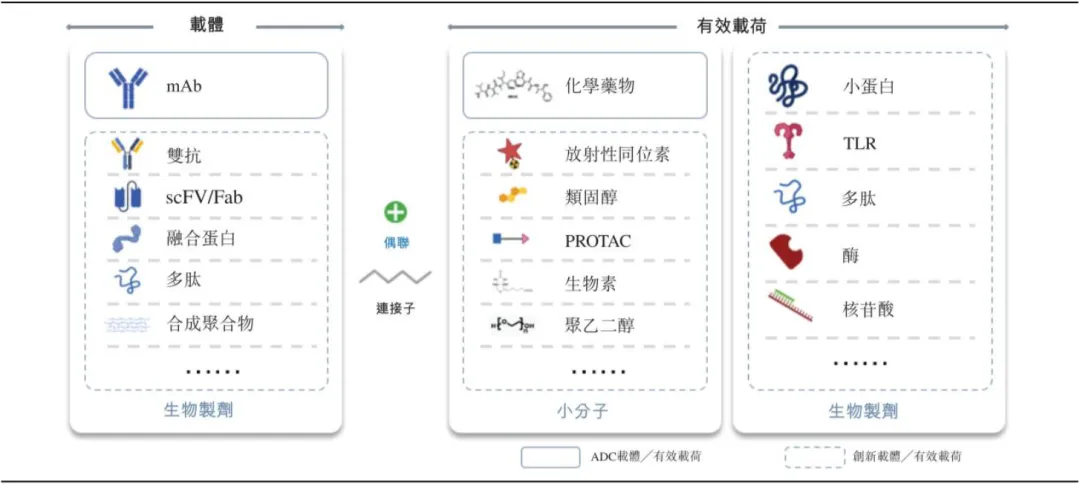
未来,随着结构生物学的突破、新型偶联技术的赋能(如万物皆可偶联的XDC平台,其中X代表载体、D代表有效载荷、C为偶联物),以及人工智能在连接子与载荷匹配上的深度参与,ADC 及其衍生药物必将突破传统大分子与小分子药物的物理与生物学天花板。这枚精密的“生物导弹”,终将真正实现从“无差别轰炸”到“精准制导微创打击”的终极愿景,为更广泛的实体瘤患者带来深度的临床缓解与长久的生存希望,而肿瘤治疗也许有一天会像慢性病一样不再那么可怕。
1
END
1
日拱一卒不期而至
公众号|小寒书巢

