基础知识:
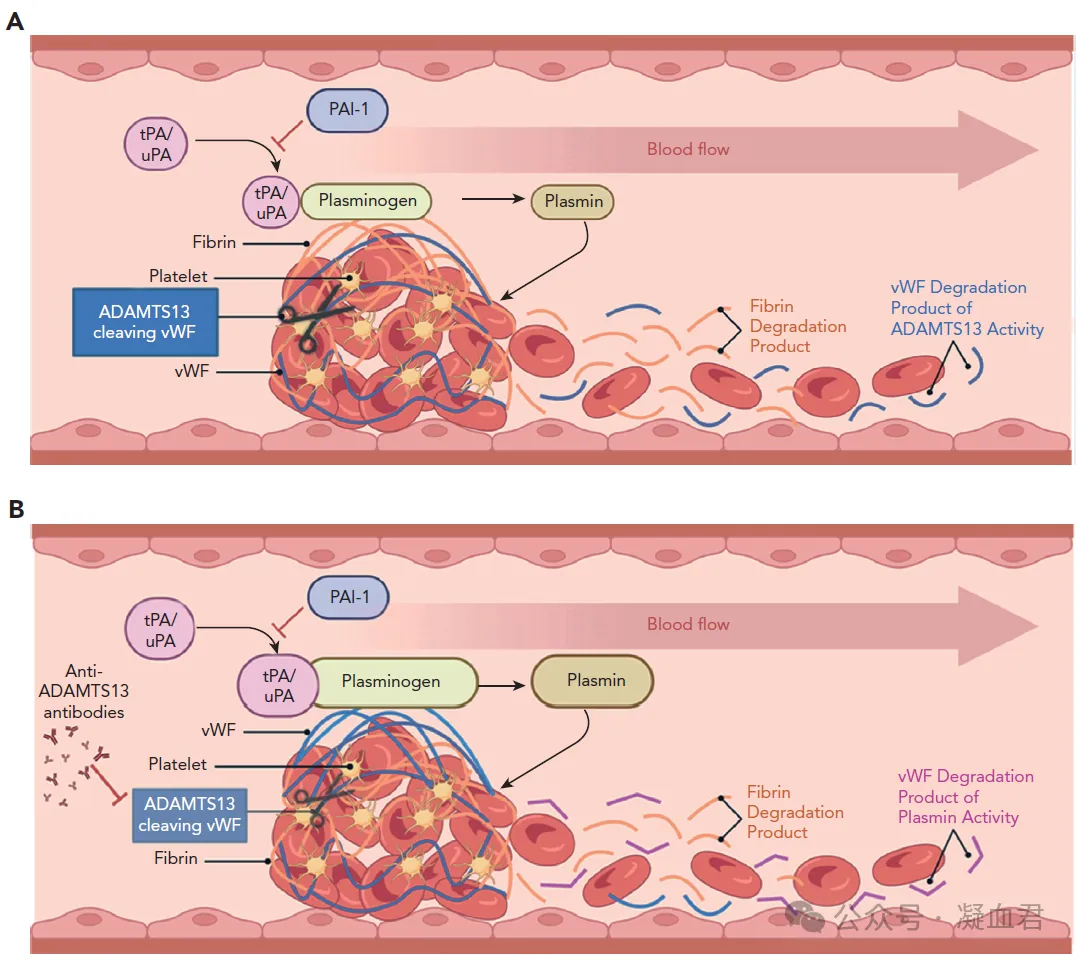
A大血管、B微血管血栓形成机制
VWF(蓝色纤维)在微血管血栓中相对丰富
而纤维蛋白(橙色纤维)在大血管血栓中相对丰富。
大血管血栓,主要由纤维蛋白降解产物(橙色)和 VWF 的 ADAMTS13 降解产物组成。
微血管血栓,降解产物中富含纤溶酶衍生的 VWF 降解产物 (cVWF)。
ADAMTS13属于血浆蛋白酶,为一种裂解血管性血友病因子(von Willebrand factor, VWF)的蛋白酶。ADAMTS13主要由肝星状细胞合成,也可由内皮细胞和巨核细胞合成。
VWF分子由内皮细胞合成并分泌至血浆,但仍附着于内皮表面,正常情况下,ADAMTS13将超大VWF分子裂解为较小多聚体,可阻止超大多聚体积聚,尤其是在剪应力较高的区域(如,较小微动脉和毛细血管)。
剪应力导致较大VWF多聚体发生构象改变,使ADAMTS13裂解部位暴露。蛋白酶活性降低时,超大VWF多聚体聚积于内皮表面,这是血小板附着和积聚处。
TTP发病机制
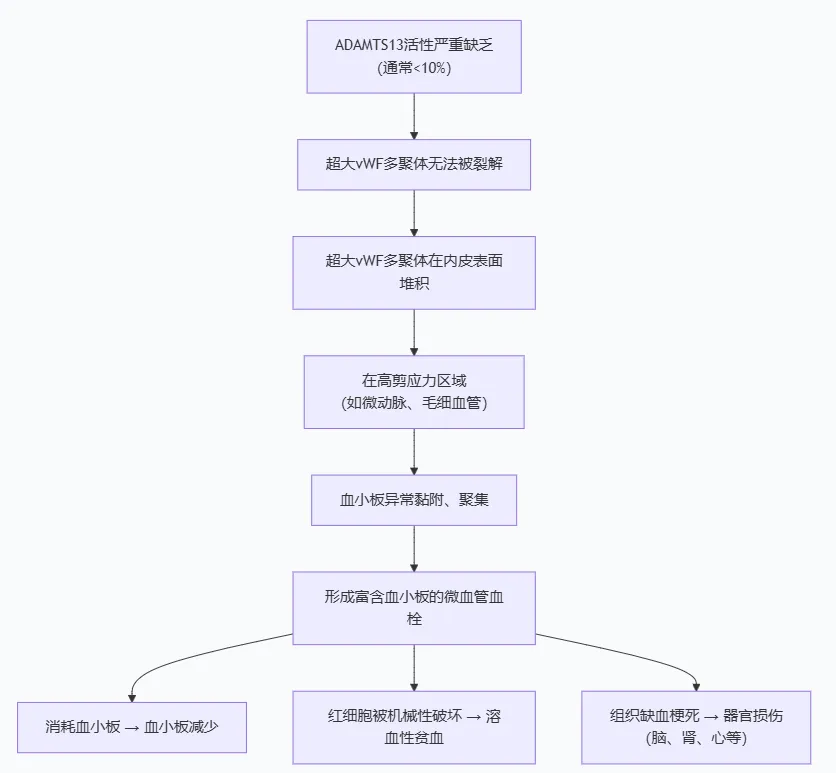
核心缺陷:ADAMTS13活性缺乏(通常降至<10%)
后果:剪刀失效,超大vWF多聚体堆积在内皮表面,尤其是在血流剪切力高的微动脉和毛细血管区域。
活性轻度降低(如肝病、脓毒症中)不足以引发TTP。
缺乏的两大根本原因
获得性(免疫性,占约95%):大多数(约95%)TTP为免疫性,由ADAMTS13抑制性自身抗体导致。
体内产生抑制ADAMTS13活性的自身抗体。这些抗体主要攻击ADAMTS13蛋白的特定结构域(如富含半胱氨酸的间隔区),直接抑制其酶活性或加速其在体内被清除。
其他情况也可能降低ADAMTS13活性,包括脓毒症、心脏手术、胰腺炎和肝病,但这种ADAMTS13水平不太可能引起TTP。
ADAMTS13活性在妊娠中期和晚期似乎也有下降,在36-40孕周及产褥期早期降至最低。
遗传性(先天性,罕见):ADAMTS13基因发生双等位基因致病变异,导致合成的蛋白酶功能缺陷或数量不足。
从缺陷到疾病的必要诱因
关键认知:仅有ADAMTS13严重缺乏并不足以立即引起TTP急性发作。
诱因作用:触发事件
这些诱因(如感染、妊娠、手术、创伤、炎症等)会刺激血管内皮细胞大量释放储存的vWF,瞬间加剧了“剪刀”的不足,从而打破平衡,引发急性血栓风暴。
血栓形成的分子机制
在剪应力高的区域,堆积的超大vWF多聚体发生构象改变,暴露出与血小板结合的部位。
血小板通过其表面的受体(如GPIbα)牢牢结合在这些超大vWF上,进而活化、聚集,形成富含血小板的微血管血栓,堵塞血管。
治疗启示:
获得性TTP治疗核心是血浆置换(清除自身抗体、补充正常ADAMTS13)联合免疫抑制(如糖皮质激素、利妥昔单抗)。
新药卡拉西珠单抗(Caplacizumab)通过直接阻断vWF与血小板的结合,能快速起效,这反过来也验证了vWF-血小板相互作用在发病中的中心地位。
遗传性TTP:预防性输注血浆或重组ADAMTS13是有效策略。
补体介导性TMA(CM-TMA,传统上称为非典型HUS)
的发病机制核心是补体替代途径的过度活化,导致内皮损伤和微血管血栓形成。这会触发多种过程,包括炎症、血小板活化、内皮损伤以及凝血级联反应激活。
核心机制:替代途径失控
基础缺陷(患者本身存在补体系统基因变异):患者存在补体调节蛋白(如补体因子H、I因子、MCP、B因子、C3等)的基因变异,或产生抗补体因子H自身抗体,导致替代途径的“刹车”失灵。
二次打击模型:基因变异仅构成易感性,多数患者需要触发因素(感染、妊娠、炎症等)诱发补体扩增,才会发病。基础失调越严重,所需触发因素越弱。
补体过度激活的后果:直接损伤血管内皮细胞(肾脏尤其敏感)。
激活血小板、促进炎症反应、启动凝血级联。
与血管性血友病因子(VWF) 相互作用:内皮表面超大的VWF多聚体可作为补体替代途径的激活平台,进一步放大损伤。
志贺毒素诱发性TMA(ST-HUS)的发病机制可概括如下:
直接细胞损伤:志贺毒素(由大肠杆菌O157:H7等菌株产生)直接损伤肾小球血管内皮细胞、足细胞及肾小管上皮细胞。
多通路激活:毒素进一步激活炎症反应和凝血级联,促进微血管血栓形成。
补体参与:部分患者可出现补体系统激活增强,放大内皮损伤和血栓形成。
该病多见于儿童,感染是首要触发因素,典型表现为溶血性贫血、血小板减少和急性肾损伤三联征。