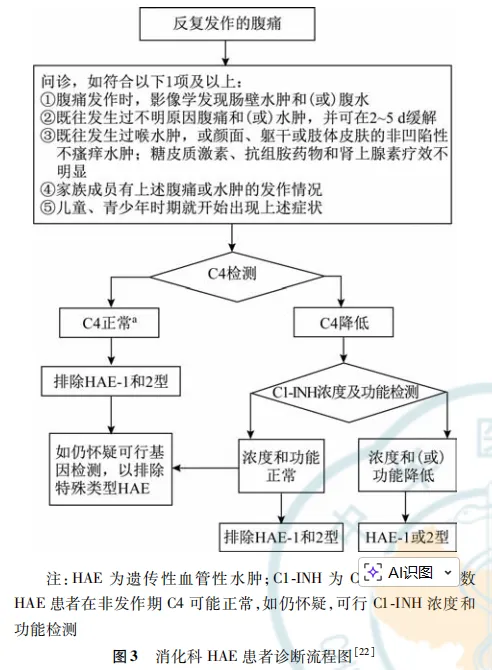
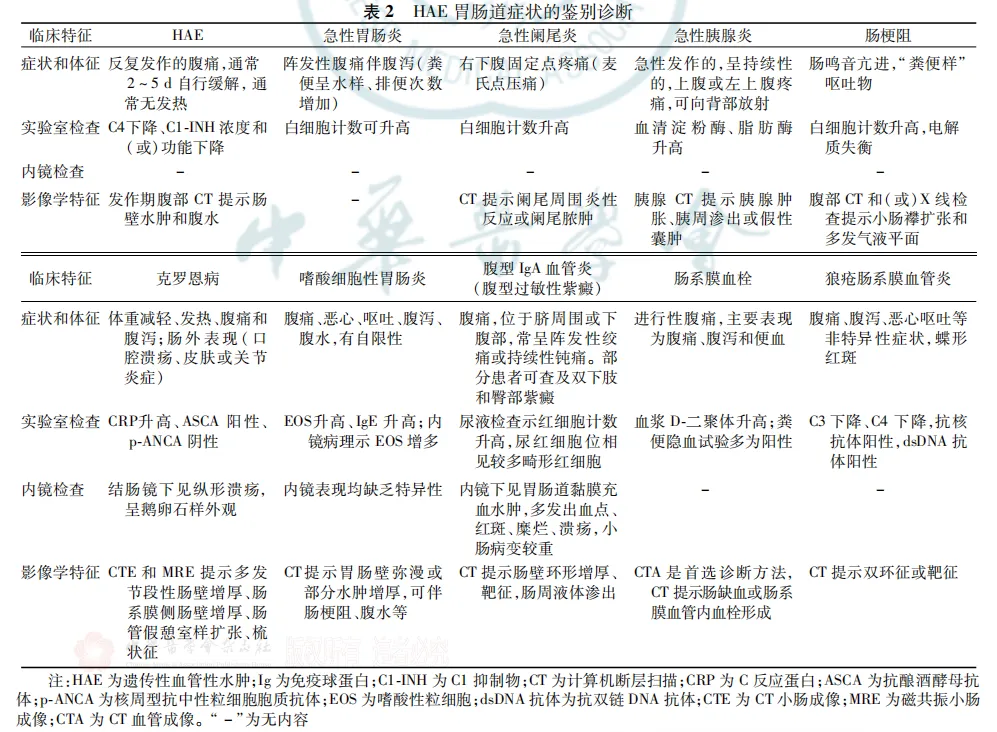
学习笔记:遗传性血管性水肿(HAE)消化科诊疗路径
定义:HAE是一种常染色体显性遗传病,主要表现为反复发生的皮肤和黏膜水肿,可累及四肢、颜面、生殖器、呼吸道、胃肠道黏膜。流行病学:全球患病率约为1:50000,属罕见病,性别、种族间无差别。分型:分三个亚型:HAE-1型最常见、HAE-2型、C1抑制物正常的HAE。HAE-1型、HAE-2型的病因是位于11号染色体上丝氨酸蛋白酶抑制家族G成员1(SERPING1)基因突变,导致体内C1抑制物(C1-INH)浓度或功能下降,影响激肽释放酶-激肽系统(KKS),导致激肽释放酶激活失控,引起缓激肽生成增加,毛细血管通透性增强,最终导致水肿的发生。C1抑制物正常的HAE(HAE-nC1-INH)的病因是由于凝血因子XI基因突变、血管生成素-1基因、纤溶酶原基因、激肽原-1重链基因、肌铁蛋白基因,以及硫酸乙酰肝素-氨基葡萄糖3-0-磺基转移酶6基因突变导致。因人而异。水肿发作频率一年数次到数十次,2-5天自然缓解,15-30%的患者可同时出现多部位水肿。诱因:大部分患者发作无明确诱因。心理压力、情绪应激、过度疲劳、外伤、手术操作、含雌激素的口服避孕药、雌激素替代疗法药物、含血管紧张素转换酶抑制剂(ACEI)成分的降压药等。胃肠道表现:肠壁水肿导致不同程度的胃肠道绞痛、恶心、呕吐、腹泻、便秘皮肤水肿:可发生在面部、四肢、躯干,表现为皮肤肿痛,无风团,非瘙痒性,非凹陷性。(注:血管性水肿主要分为肥大细胞介导的、特发性、缓激肽介导的三类,肥大细胞介导的皮肤血管性水肿多伴有风团和瘙痒)家族史:绝大部分为常染色体显性遗传,50%的子代会遗传。25%HAE为新发的基因突变。(注:补体C4水平下降的原因:C1抑制物缺乏导致C1蛋白自身活化、裂解并消耗大量C4蛋白。无论在发作期或缓解期,C4水平均下降,筛查的灵敏度为81-96%。补体C3水平正常。狼疮肠系膜血管炎C3、C4均下降。)基因检测:更有助于确诊C1抑制物正常的HAE(HAE-nC1-INH)影像学检查:腹部CT或B超可见腹水,CT可观察到节段性肠壁或黏膜水肿及环周增厚(最常累及空肠和十二指肠),肠系膜血管增多、增粗。(注:克罗恩病CT表现为肠系膜侧肠壁增厚)- 一线药:艾替班特(通过选择性和竞争性拮抗缓激肽B2受体发挥作用),2021年在中国上市,用于成人、青少年和≥2岁儿童的HAE急性发作,30mg/次,腹部皮下注射,如症状未缓解或复发,间隔6小时再次用药,24小时不超过3次。(血源性C1-INH、重组 C1-INH、艾卡拉肽均未在中国大陆地区上市)
- 胃肠道症状对症用药:丁溴东蓢菪碱、甲氧氯普胺、丙氯拉嗪治疗绞痛、恶心、呕吐
预防性治疗:对于已明确诊断的患者均推荐长期预防治疗,减少发作- 国内一线药:拉那利尤单抗(单克隆抗体药物,用于≥12岁患者,靶向抑制HAE患者体内活化的血浆激肽释放酶)
- 儿童用药:抗纤溶制剂(氨甲环酸,有学者提出此观点,机制不明)
- 国外还有的药:血源性C1-INH(cinryze)、贝罗司他
从消化科临床视角重点总结:绝大部分HAE会出现胃肠道症状,常被误诊为急腹症,甚至导致不必要的外科手术,所以提高消化科医师对HAE的认知尤为重要,临床上碰到不明原因反复发作的腹痛、恶心、呕吐、腹泻伴或不伴皮肤水肿,常规治疗效果不佳,需考虑HAE。声明:以上为镜途慢漫阅读文献《遗传性血管性水肿消化科诊疗路径》的学习笔记,如有错误,望同行指正,不胜感激。