各位小伙伴大家好啊~4月的CNS又刷屏了。我们花时间对几篇拆解了一下,以下是精简后的文献笔记,话不多说直接开始!
Science | 以癌攻癌:让肿瘤反戈一击
标题:Turning tumors against themselves
通讯: Fábio F. Rosa(瑞典隆德大学)
一句话总结:
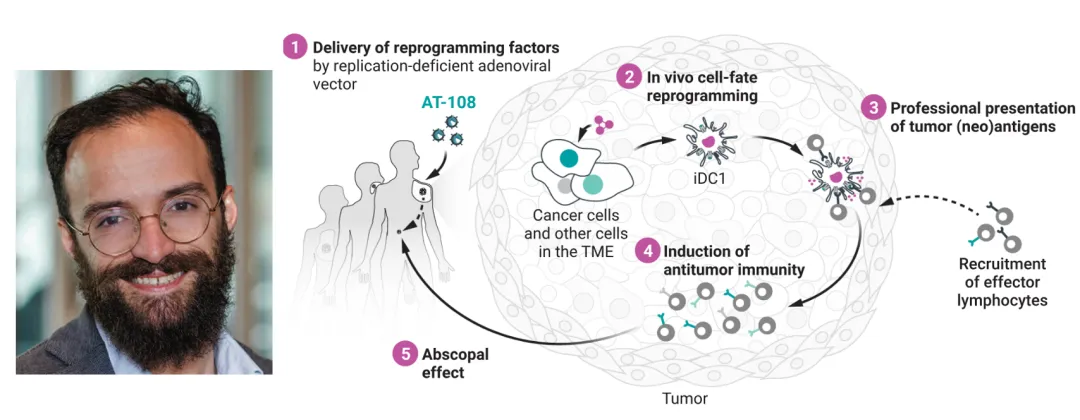
强调一下这篇并不是research,而是一篇2026年《Science》创新大奖得主的获奖Essay,它复盘了作者Fábio Rosa十年磨一剑的历程:从2018年通过筛选18个树突状细胞相关转录因子,锁定PU.1、IRF8、BATF3(PIB)三个因子,首次将小鼠和人类成纤维细胞直接重编程为功能性1型常规树突状细胞(cDC1),具备抗原交叉呈递能力。到2024年将PIB三因子装进腺病毒载体瘤内注射,在小鼠体内把肿瘤细胞直接重编程为cDC1样细胞,使其暴露自身肿瘤抗原并重塑微环境,在黑色素瘤等多种模型中实现原位肿瘤消退和远端转移灶清除(远隔效应)。
当课题的疗法涉及复杂的体外细胞操作,可以尝试:能不能换一种递送介质(AAV病毒、腺病毒、脂质体LNP),在体内实现靶向修改? In vivo 是未来十年的主线,符合临床落地的逻辑。2018年发基础论文,同步申请专利,成立Asgard Therapeutics;2024年发应用论文,同步融了3000万欧元A轮,科学故事和公司故事并行推进。
Cell | CAR-T一箭双雕,连根拔起实体瘤老巢
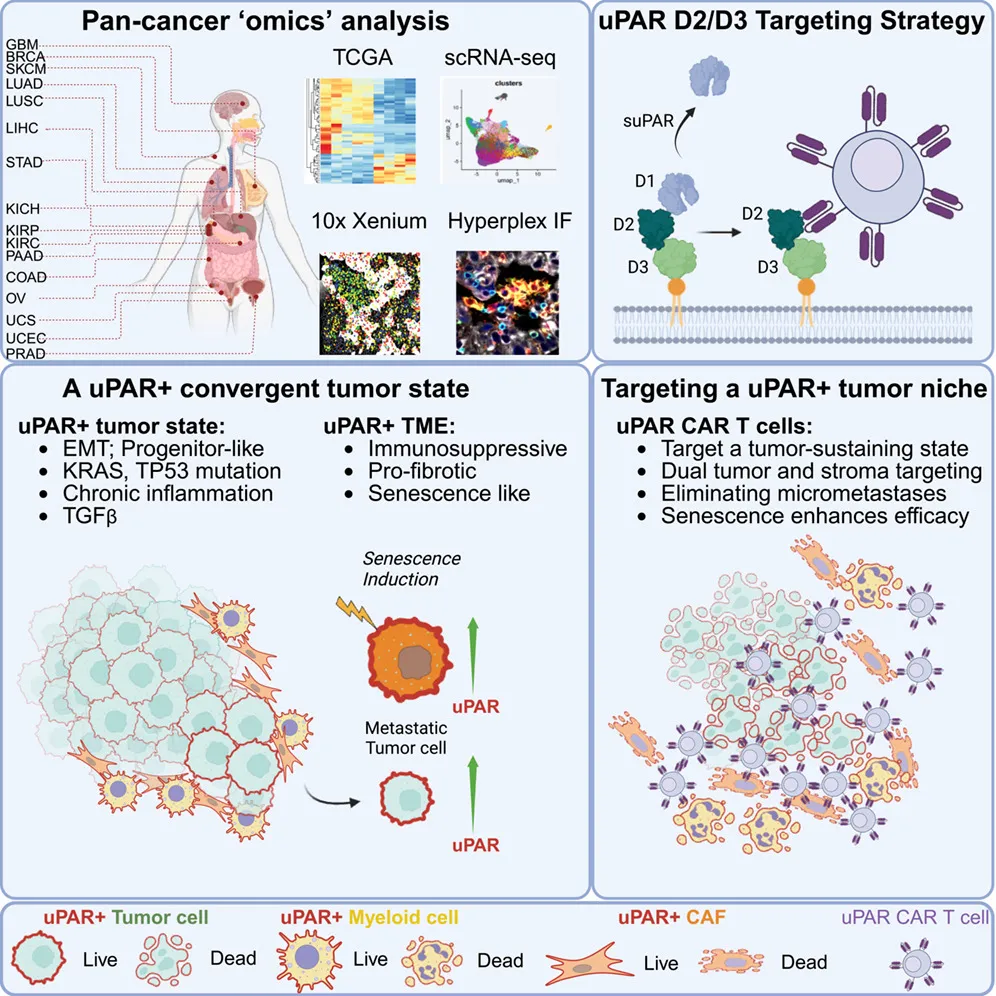
标题:A convergent uPAR-positive tumor ecosystem creates broad vulnerability to CAR T cell therapy
通讯: Scott W. Lowe (纪念斯隆-凯特琳癌症中心)
一句话总结:
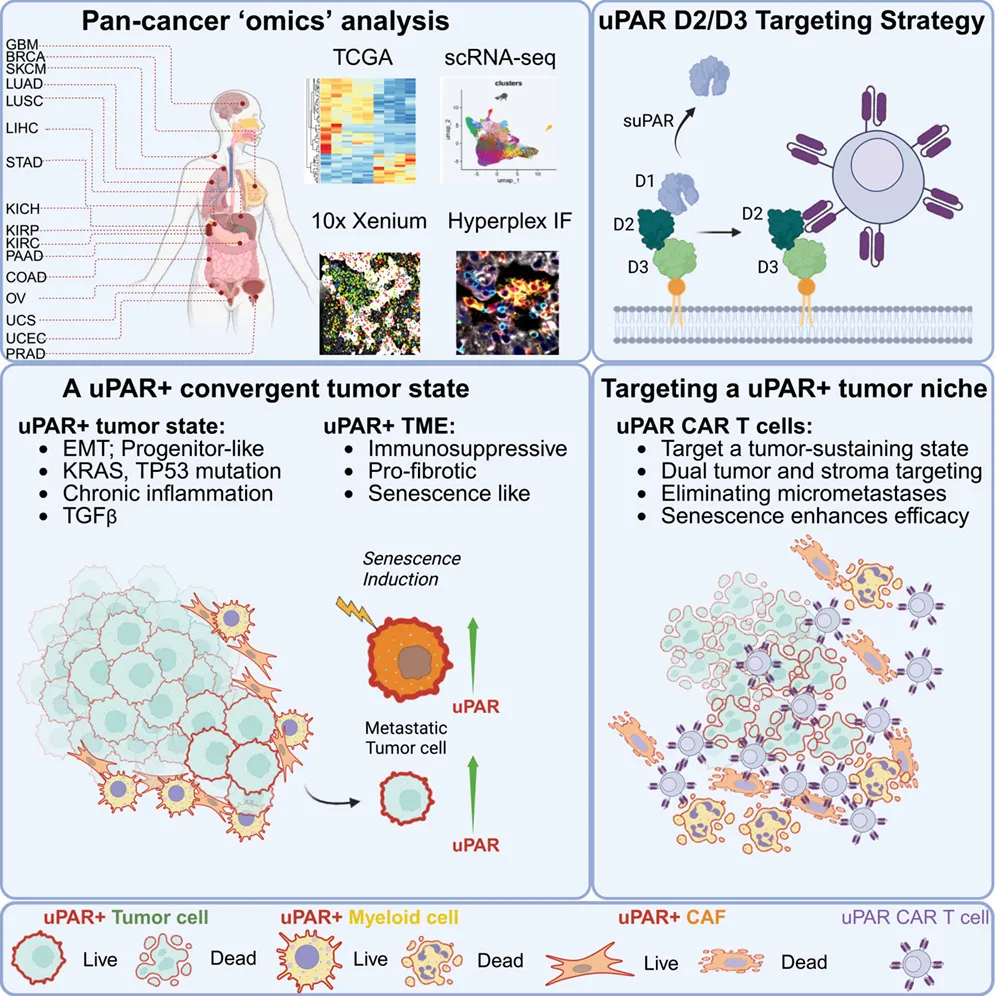
这项研究发现uPAR在p53突变的实体瘤中广泛高表达,还广泛表达于促纤维化、免疫抑制的衰老基质细胞上;据此开发的uPAR CAR-T细胞能实现肿瘤+微环境双杀,且通过联合传统化疗药物(诱导细胞衰老以提高靶点表达)可大幅提升疗效。
这里面有个很讨巧的部分是,既然正常髓系细胞也有uPAR,为什么小鼠没死于骨髓抑制?作者是测一下造血干细胞/祖细胞,发现它们不表达这个靶点。只要祖细胞还在,成熟细胞被杀光了也能很快再生。所以在脱靶毒性问题上这个解释还是自洽的。此外,uPAR有脱落形式(suPAR),即uPAR容易从细胞表面脱落(shedding)成游离态,但作者筛的是D2-D3结构域的抗体,绕开了最容易脱落的D1,避免了分泌型靶点抗原对CAR-T的消耗和干扰。做膜蛋白靶点要前期查清结构域,筛抗体时针对性地选不同的区段。
Nat Genet | 反向操作破局T细胞杀伤抗性
标题:High-content CRISPR activation screens identify synthetically lethal RNA-based mechanisms to sensitize cancer cells to targeted T cell cytotoxicity
通讯: Livnat Jerby(斯坦福大学医学院)
一句话总结:
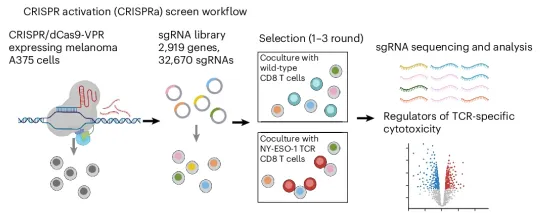
本研究另辟蹊径地利用CRISPR激活(CRISPRa)筛选和首创的原位Perturb-seq(结合空间转录组)技术,鉴定出一系列能选择性增敏癌细胞对TCR特异性T细胞杀伤的RNA可表达基因(如CASP3、SAFB、MYC、WNT配体等),能在不影响癌细胞基础存活率的前提下,特异性放大T细胞的杀伤作用,提出了免疫RNA合成致死概念。
本文又有一个概念包装:传统的“合成致死”通常指两个基因同时突变或失活才会导致细胞死亡(如经典的BRCA与PARP抑制剂)。本文作者把这个概念进行了延展:癌细胞平时过表达这些靶点(如 CASP3)活得好好的,但只要一遇到T细胞攻击,就会瞬间死亡。这种平时无毒,遇敌即死的机制被包装成免疫RNA合成致死 (immune RNA-based synthetic lethality)。传统敲除筛选只能找抑制了会怎样,而激活筛选直接找过表达就能增敏的基因。这类靶点本身就是RNA药物的天然候选,转化路径比敲除靶点更短更直接。
Cell | 癌基因点燃脑内炎症
标题:Somatic cancer variants enriched in Alzheimer's disease microglia-like cells drive inflammatory and proliferative states
通讯: Christopher A. Walsh(波士顿儿童医院和哈佛医学院)
一句话总结:
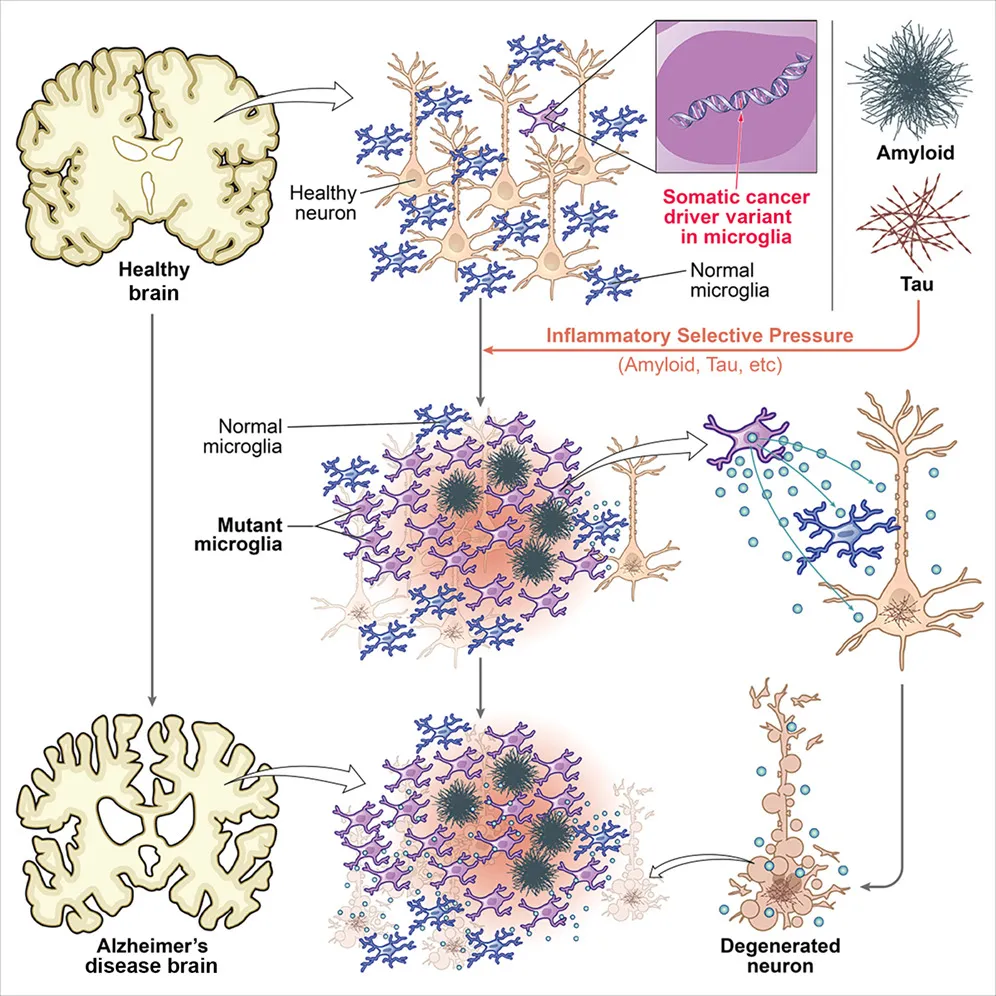
本文借公共队列与公开数据、FANS分选+扩增子测序等发现阿尔茨海默病患者大脑中的小胶质细胞样脑巨噬细胞(MLBMs)富集了与克隆性造血相关的癌症驱动基因体细胞突变,这些突变促使细胞转向促炎、增殖的疾病相关状态,从而可能推动神经退行性变。
文章创新处在于将经典的肿瘤学概念(体细胞突变、克隆造血、肿瘤抑制基因失活)引入了神经退行性疾病。成熟的肿瘤靶向机制(如文中提到的PI3K-PKB/Akt通路或表观遗传调控因子TET2/DNMT3A)有潜力作为新靶点,切入到其他免疫或炎症介导疾病中。体外验证这部分,通过CRISPR构建了三对关键基因(DNMT3A, TET2, ASXL1)的等基因iPSC系,并分化为小胶质样细胞,干净利落地证明了突变直接驱动促炎表型。
Nature | CAR-T一箭双雕,连根拔起实体瘤老巢
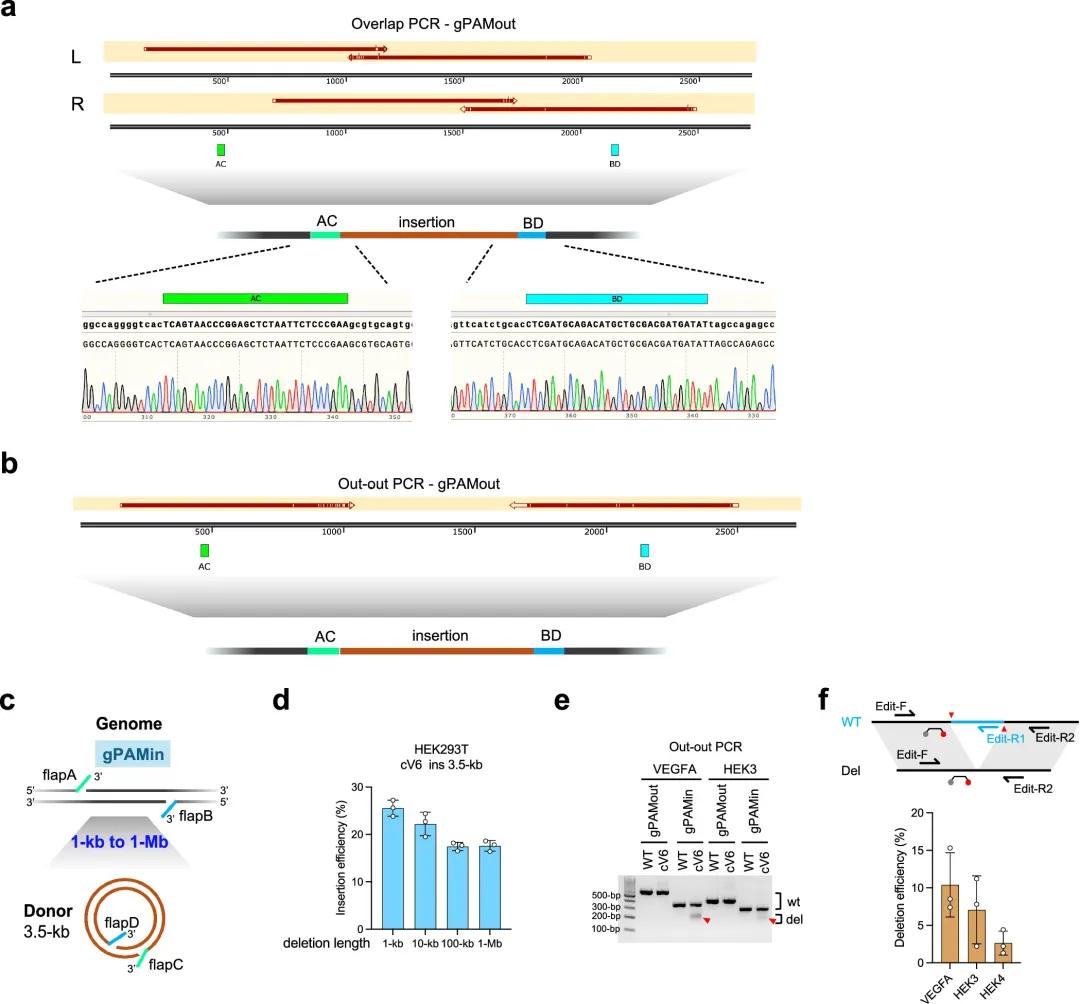
标题:Quadruple pegRNA enables programmable and efficient large genomic insertion
通讯: 张楹(Ying Zhang) 与 殷昊(Hao Yin)武汉大学(泰康生命医学中心/医学研究院)
一句话总结:
本研究开发了一种名为QuadPE的新型先导编辑系统,通过使用四条经设计的向导RNA(pegRNA)分别靶向基因组和供体DNA,在不产生双链断裂且不依赖外源整合酶或转座酶的情况下,实现了大片段DNA(高达26 kb)在分裂和非分裂细胞(如神经元和T细胞)中的高效、精准插入。
在分裂活跃的细胞里,做简单的基因敲入可以用带抗性的质粒筛稳转株,做复杂一点的有现成的慢病毒、转座子系统。不过一些难搞的细胞原代神经元什么的可以试试。
Cell | CRISPR全基因组筛选重塑原代T细胞的HIV宿主互作图谱
标题:Systematic discovery of pro- and anti-HIV host factors in primary human CD4+ T cells
通讯: Alexander Marson 和 Ujjwal Rathore(加州大学旧金山分校和格拉德斯通研究所)
一句话总结:
本研究在原代人类 CD4+ T 细胞中进行了首个全基因组范围的 CRISPRa(激活)和 CRISPRn(敲除)双向筛选,系统鉴定出数百个促 HIV 感染和抗 HIV 感染的宿主因子,并揭示了新型强效抗病毒蛋白 PPID(通过结合 HIV 衣壳阻断其入核)和 PI16(通过干扰肌动蛋白重塑阻断病毒融合)的作用机制。
在筛选出大量未知基因后,怎么快速对它们的功能阶段进行分类?作者用了一个低成本的对照:VSV-G 假型 HIV 病毒(它通过内吞进入细胞,不依赖 HIV 传统的包膜受体)。如果一个基因在野生型 HIV 筛选里是 Hit,但在 VSV-G HIV 筛选里失效了,说明它作用在病毒进入(Entry)阶段;如果它在两者中都是 Hit,说明它作用在进入后(Post-entry)的阶段。这种利用不同包膜/侵入机制的病毒株进行比对的思路值得借鉴的。
好啦,本期 4月顶刊学习笔记到这里就结束啦~欢迎大家点赞、收藏、转发,我们下月继续分享高分好文,不见不散!
文章著作权归文章作者所有,
未经允许禁止转载!
咨询和合作:
cnsmaker@126.com