Bioassay validation is in some respects similar to validation of other analytical methods. Key among the similarities is demonstration of fitness for use. This extends from bioassay design and development and is continually managed throughout the bioassay life cycle. For purposes of this chapter,
will mean that the bioassay method, and the procedures that are developed using the method, are capable of supporting key biological product decisions. In the case of a release procedure, for instance, performance characteristics should be such as to limit the risks of making a poor release decision [e.g., out of specification (OOS)]. In this regard, the release specification is essential to defining the performance requirements for that use.
在某些方面,生物检定验证与其他分析方法的验证具有相似性,其中最核心的共同点是证明适用于预期用途。这一要求贯穿于生物检定的设计、开发,并在其整个生命周期中持续实施与管理。就本章而言,适用于预期用途是指:生物检定方法以及基于该方法建立的分析程序,能够支持对生物制品的关键决策。例如在放行程序中,方法的性能特征应能降低做出错误放行决策的风险(如结果超标 OOS)。在这一意义上,放行标准是定义该用途下性能要求的关键依据。
It is expected that the design of the bioassay method and associated suitability criteria have been completed prior to implementation of the validation and documented in the SOP. This includes but is not limited to identification and alignment of key structural factors, e.g., plates and plate layout in a cell-based bioassay and cages and cage position in an in vivo method. Robustness (or optimization) studies should have been carried out to establish ranges on key factors such as incubation temperatures and times, and ages and weights of animals. Those ranges should be a part of the SOP and adhered to during the validation.
生物检定方法的设计及相关适用性标准,应在验证实施前完成,并在标准操作规程(SOP)中形成文件。这包括但不限于关键结构要素的识别与统一,例如细胞法生物检定中的培养板及布局、体内方法中的动物笼具及笼位等。应已开展 ** 耐用性(或优化)** 研究,以确定关键条件的范围,如孵育温度与时间、动物年龄与体重等。这些范围应纳入 SOP,并在验证期间严格遵守。
Suitability criteria should be proposed as part of the bioassay SOP, while a provisional release procedure format is used for calculation of validation acceptance criteria. This may be updated according to findings during the validation. Any update should be directed, however, towards improving the performance or efficiency of the procedure. Suitability criteria may be updated, including elimination, when there is sufficient experience to determine the long-term impact on bioassay control. The provisional release procedure format should be changed immediately after validation when it has been determined that this is insufficient to deliver an adequately precise RV, while further changes can occur after sufficient experience with the procedure.
适用性标准应作为生物检定 SOP 的一部分进行拟定,同时采用暂定放行程序模式用于计算验证可接受标准。该模式可根据验证中的发现进行更新,但任何更新均应以提升程序性能或效率为导向。在积累足够经验、能够判断对生物检定控制的长期影响后,适用性标准可进行更新(包括删减)。若经确认暂定放行程序模式无法提供精密度满足要求的可报告值(RV),则应在验证后立即修改;后续进一步调整可在获得足够程序使用经验后实施。
The use of corrective action and information coming from continued performance verification is particularly important when statistical criteria (e.g., equivalence and noninferiority testing) are used to assess conformance of validation parameters to their acceptance criteria. This is because the validation study may not be powered sufficiently to verify satisfactory bioassay performance. This also reflects the need to be vigilant to bioassay performance throughout its life cycle.
在采用等效性、非劣效性检验等统计标准评估验证参数是否符合可接受标准时,纠正措施的应用以及来自持续性能确认的信息尤为重要。这是因为验证研究的统计效力可能不足以充分证明生物检定性能满足要求,也体现了在方法整个生命周期中持续关注其性能的必要性。
The bioassay validation design should include important ruggedness factors with as many levels as is practically feasible. Introduction of factors should take precedence over replication of the procedure (i.e., replication of the provisional release procedure format) to optimize the use of resources and to best represent the impact of changes in these factors on long-term performance of the method. Attention to factors rather than release procedure format provides information to verify the provision release procedure and to effectively design other procedures using the bioassay. Factors that have been resolved during development may be included during validation when these are critical or may interact with another factor (e.g., temperature may be re-explored if the validation includes a heat sensitive reagent). The design of the bioassay validation should likewise acknowledge the range of potencies that will be encountered during routine release, as well as during long-term and accelerated stability evaluations.
生物检定验证设计应纳入重要的稳健性因素,并在实际可行前提下设置尽可能多的水平。引入这些因素应优先于对程序的重复(即对暂定放行程序模式的重复),以优化资源利用,并更真实地反映这些因素变化对方法长期性能的影响。关注因素而非放行程序模式,可用于验证暂定放行程序,并有效设计基于该生物检定方法的其他程序。在开发阶段已确定的因素,若属于关键因素或可能与其他因素存在交互作用,可在验证阶段重新纳入考察(例如,验证中使用热敏试剂时可重新考察温度)。生物检定验证设计同样应覆盖日常放行以及长期、加速稳定性研究中可能遇到的效价范围。
In bioassay validation,
refers to the relationship between observed and expected log potencies rather than between signal and dose (i.e., dose response). Their regression (log observed versus log dose) should ideally have a slope equal to 1.0. Any deviation from a unit slope indicates proportional bias (i.e., bias between results across a range). Proportional bias has an impact on comparisons of potency results across a range (e.g., between stability time points or in stability slope, between results before and after a process change, etc.).
在生物检定验证中,线性是指观测效价对数值与预期效价对数值之间的关系,而非信号与剂量(即剂量–响应)之间的关系。其回归方程(观测对数值 vs 剂量对数值)理想斜率应等于 1.0。任何偏离单位斜率的情况均表明存在比例偏倚,即不同效价水平间测定结果的偏倚。比例偏倚会影响不同水平下效价结果的比较(例如稳定性考察各时间点之间、稳定性斜率之间、工艺变更前后结果之间等)。
Relative accuracy and linearity should ideally be prospectively addressed during bioassay design and development. Strategies such as randomization should be incorporated into the bioassay method to mitigate the impacts of factors that can generate bias in relative potency determination or comparisons. Steps in relative potency determination, such as similarity testing, can have a direct impact on RB or nonlinearity of potency determination. Thus, similarity acceptance criteria should be established during development to minimize these impacts prior to validation.
相对准确度与线性宜在生物检定设计与开发阶段进行前瞻性控制。应将随机化等策略纳入生物检定方法,以降低可能导致相对效价测定或比较出现偏倚的因素影响。相对效价测定中的相关步骤(如相似性检验)会直接影响相对偏倚(RB)或效价测定的线性。因此,应在开发阶段建立相似性可接受标准,在验证前将这些影响降至最低。
Determination of IP is perhaps the most important goal of bioassay validation, particularly if relative accuracy and linearity have been successfully addressed during bioassay design and development. The validation design should support the estimation of VCs associated with both within-run and between-run sources of variability or of meaningful ruggedness factors such as analysts and reagent lots. Reliable estimates of VCs can be used either to verify that the provisional release procedure format meets the precision requirement in the ATP or to update the format, as well as to design other procedures using the bioassay method (e.g., standard qualification, stability studies).
中间精密度(IP)的确定是生物检定验证中最重要的目标之一,尤其当相对准确度与线性已在设计与开发阶段得到良好控制时。验证设计应支持对批内、批间变异来源以及分析员、试剂批次等重要稳健性因素对应的 ** 方差成分(VC)** 进行估算。可靠的方差成分估算可用于:验证暂定放行程序模式是否满足分析目标概况(ATP)中的精密度要求、更新程序模式,以及设计基于该生物检定方法的其他程序(如标准品标定、稳定性研究)。
CIs should be calculated for the validation parameters using methods described in
Analytical Data—Interpretation and Treatment 〈1010〉
and Burdick and Graybill (
). Conformance of validation parameters to their acceptance criteria should be carried out using either equivalence or noninferiority testing when the validation has been designed with sufficient power to perform a statistical assessment. Failure to satisfy the noninferiority criterion for IP may result in a change to the procedure format.
应按照《分析数据 —— 解析与处理》〈1010〉及 Burdick 与 Graybill(1)所述方法计算验证参数的置信区间(CI)。若验证设计具备足够统计效力,应采用等效性或非劣效性检验判定验证参数是否符合可接受标准。若中间精密度不满足非劣效性标准,可能需要对程序模式进行修改。
The remainder of this section will discuss bioassay validation design, the bioassay validation protocol, bioassay validation data analysis, documentation of bioassay validation results, and continued performance verification including bioassay maintenance.
本节后续内容将论述生物检定验证设计、生物检定验证方案、验证数据分析、验证结果文件化,以及包括生物检定维护在内的持续性能确认。
2.1 Bioassay Validation Design 生物检定验证设计
The bioassay validation should include samples supporting the range of the product specification and extended to ensure the reliability of each run used to determine the RV for a test article. Alternatively, the release procedure can include the provision to repeat the series of runs using alternative doses when some runs fail suitability. The bioassay range represents the extreme potency levels that show acceptable relative accuracy, IP, and linearity. Additional consideration should be given to the extended range of real-time and accelerated stability results.
生物检定验证应采用覆盖产品质量标准范围的样品,并可适当扩展,以确保每一次用于计算供试品可报告值(RV)的运行结果均可靠。作为替代方案,放行程序可规定:若部分运行未通过适用性要求,可采用其他剂量重新进行系列运行。生物检定的范围是指能够满足相对准确度、中间精密度与线性要求的极端效价水平。同时还应考虑实时与加速稳定性结果所覆盖的扩展范围。
In order to achieve representative estimates of relative accuracy, validation samples should be produced from the standard material (or the source of the standard material). This is to reduce the impact of the uncertainty of measurement of another sample on the estimate of bias. A minimum of three potency levels should be included in the validation, while five levels are recommended for a reliable assessment of linearity and to mitigate the risk of a restricted bioassay range (i.e., due to failure at one or the other of the validation sample extremes).
为获得具有代表性的相对准确度估算,验证样品应采用标准品(或标准品原料)制备,以降低其他样品的测量不确定度对偏倚估算的影响。验证中应至少包含三个效价水平,推荐采用五个水平,以便可靠评估线性,并降低因验证样品某一极端水平不合格而导致生物检定范围受限的风险。
It is expected that bioassay development (as detailed in 〈1032〉) will yield a method design based on knowledge of the within-run factors that affect the assessment of similarity and measurement of potency. The design of the validation should reflect knowledge of the long-term (between-run) factors that might influence the measurement of potency. It is helpful to include all sources associated with large variations and those where the effects of a factor are poorly understood. During development, a laboratory might alternatively employ “robust design” (
), which is a strategy that optimizes settings of within-run factors while simultaneously minimizing the variability due to between-run factors.
预期生物检定开发(详见〈1032〉)将基于对影响相似性评价与效价测定的运行内因素的认知,形成方法设计。验证设计则应体现对可能影响效价测定的长期(运行间)因素的认知。纳入所有变异较大的来源以及作用尚不明确的因素是有益的。在开发阶段,实验室可选择采用 “稳健设计”(2),即在优化运行内因素设置的同时,最小化运行间因素带来的变异。
A release procedure format that results in RV for a test material may be specified during development and used to test clinical materials. This format may be provisional owing to incomplete knowledge about the impacts of factors that may impact bioassay variability over its life cycle. During validation, IP is studied using combinations of factors that are formulated into validation runs. Validation runs should be conducted the same as bioassay method runs to be representative. Factors may be incorporated into validation runs using strategies that alter these in a planned way.
在开发阶段可规定能够得出供试品可报告值(RV)的放行程序模式,并用于临床样品检测。由于对可能影响生物检定整个生命周期变异性的因素认知尚不充分,该模式可为暂定模式。在验证阶段,通过将多种因素组合纳入验证运行,对中间精密度进行研究。验证运行的执行应与常规生物检定运行保持一致,以保证代表性。可采用计划性变更策略将因素纳入验证运行。
Using DOE, validation runs can be balanced across ruggedness factors (i.e., equal numbers of runs can be performed at all levels of the factors).
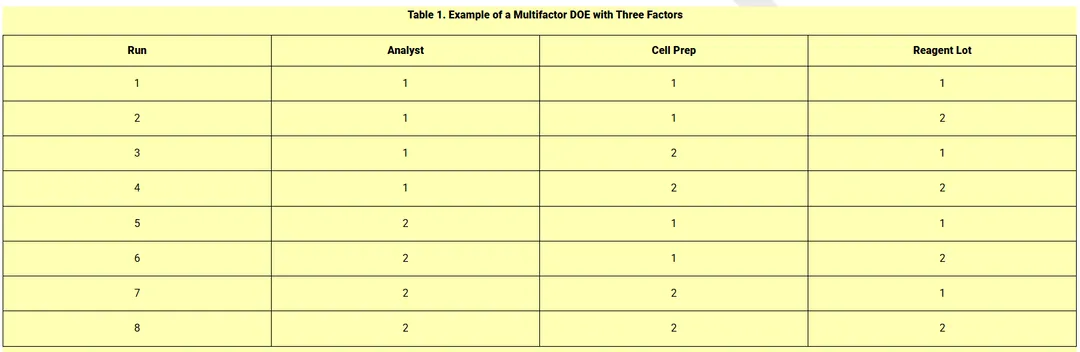
shows an example of a multifactor DOE that incorporates multiple analysts, multiple cell culture preparations, and multiple reagent lots into the validation plan.
借助试验设计(DOE),验证运行可在各稳健性因素间实现均衡设计,即在所有因素水平下开展数量相等的运行。表 1 给出了一个多因素 DOE 示例,将多名分析人员、多次细胞培养制备以及多批试剂纳入验证方案。
This represents a full factorial design where each analyst performs the bioassay with each of the 2 cell preparations and each of the 2 reagent lots. Balance is noted in having
= 4 runs at each level of the three factors in the design. Within-run replicates may be performed within some or all runs to estimate a within-run component of variability and to facilitate the design of bioassay procedures (see
5. Uses of Bioassay Validation Study Results
).
这属于全因子设计,每名分析人员均使用 2 种细胞制备物和 2 批试剂分别进行生物检定。设计中三个因素的每个水平均有 n=4 次运行,体现了均衡性。可在部分或全部运行内开展批内重复试验,以估算批内变异成分,并为生物检定程序的设计提供支持(见第 5 节 生物检定验证研究结果的应用)。
Fractional factorial designs may be employed to manage the validation study size when many factors have been identified. It is important to note that this use of DOE is not to estimate factor effects. Fractional factorial designs are used to maintain balance across factors while accommodating two-way interactions of factors.
当考察因素较多时,可采用部分因子设计控制验证研究规模。需要明确的是,此处采用试验设计(DOE)的目的并非估算各因素的效应,而是在兼顾因素间二阶交互作用的同时,保持因素水平间的均衡性。
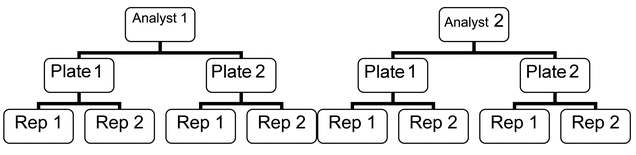
Designs using nesting (e.g., replicates nested within plate, plates nested within analyst) may likewise be used during validation. An example of a nested design is shown in
.
验证过程中同样可采用嵌套设计(例如,重复组嵌套于培养板内,培养板嵌套于分析人员内)。图 1 给出了嵌套设计的一个示例。
Figure 1. Example of a nested design using two analysts.
Components of variability can be estimated from the validation results from one or a combination of these designs. Designs using only two levels of a factor (e.g., two analysts) are insufficient to obtain a reliable estimate of its contribution to IP, while analysis of routine control data (as part of continued performance verification) should be used to support the decision to replicate a factor in the release procedure design. Nevertheless, significant sources of variability may have been identified during bioassay development. In this case, the validation should confirm their impact and their variability can be used to develop a bioassay format that meets the requirement for precision in the procedure ATP.
可通过上述一种或多种设计的验证结果估算变异成分。仅采用两个水平的因素(如两名分析人员)不足以可靠估算其对中间精密度(IP)的贡献,而对日常质控数据的分析(作为持续性能确认的一部分)应用于支持是否在放行程序设计中对该因素设置重复的决策。尽管如此,在生物检定开发期间可能已识别出显著的变异来源。在此情况下,验证应确认其影响,且这些变异可用于开发满足分析目标概况(ATP)中程序精密度要求的生物检定模式。
An additional consideration in bioassay validation design is the number of validation runs. The number of runs (and within-run repeats) is driven by the risks associated with conclusions drawn from the validation data analysis. This topic is discussed further in
2.3 Bioassay Validation Sample Size
.
生物检定验证设计中的另一项考虑是验证运行次数。运行次数(及运行内重复次数)由验证数据分析所得结论的相关风险决定。该主题将在 2.3 生物检定验证样本量中进一步讨论。
As discussed in
, it is less desirable to employ the provisional release procedure format within the design of the validation than to dedicate validation runs to inclusion and replication of important long-term sources of variability. Estimates of variability due to these components can be used to verify the precision of the provisional release procedure format or to explore alternative formats. In addition, a validation that delivers reliable estimates of long-term VCs (estimates of the variance due to individual factors) can support different procedures using the bioassay method, with incorporation of significant factors as well as various numbers of within-run and between-run replicates into their designs. This is particularly valuable because different uses of the bioassay (e.g., lot release, qualification of new standards, comparability of a new product manufacturing process, or stability studies) will have different performance requirements, requiring different designs and formats to support these.
如第 1 章引言中所述,在验证设计中采用暂定放行程序模式,不如在验证运行中专设并重复考察重要的长期变异来源。通过估算这些成分导致的变异,可用于验证暂定放行程序模式的精密度或探索替代模式。此外,若验证能提供可靠的长期方差成分(VC)估算(即各独立因素导致的方差估算),则可支持采用该生物检定方法的不同程序,将显著因素以及不同数量的运行内与运行间重复纳入其设计中。这一点尤为重要,因为生物检定的不同用途(如批次放行、新标准品标定、新产品生产工艺可比性或稳定性研究)具有不同的性能要求,需要不同的设计与模式予以支持。
This approach might be thought of as “validating the bioassay method” rather than “validating the release procedure”, and its importance is illustrated for the validation of an in vivo (animal) potency bioassay. While the final validated format for an in vivo bioassay procedure may require multiple runs to have sufficient precision in the RV, it is typically impractical to design an effective validation with the release procedure in its design.
该思路可理解为 **“对生物检定方法进行验证”,而非“对放行程序进行验证”**,其重要性可通过体内(动物)效价生物检定的验证加以说明。尽管体内生物检定程序最终经验证的模式可能需要多次运行才能使可报告值(RV)具备足够精密度,但在验证设计中纳入放行程序通常不切实际。
In fact, the burden of validating a bioassay procedure is generally related to the underlying variability of the method. A procedure for a highly variable method (in vivo, plaque count, TCID50) is likely to include multiple replicates to achieve the ATP requirement for precision of the RV. This is no less important for more precise methods where more factors and replicates of those factors can result in more robust information regarding bioassay performance.
事实上,生物检定程序的验证负担通常与方法本身的变异程度相关。高变异方法(体内法、空斑计数法、TCID₅₀)的程序可能需要设置多个重复以满足 ATP 对可报告值(RV)精密度的要求。这对于精密度更高的方法同样重要,因为纳入更多因素并对这些因素设置重复可获得更耐用的生物检定性能信息。
It is important to note, however, that to obtain a representative estimate of the IP of the method, the validation should be performed in accordance with restrictions that have been imposed during development (or as part of the method SOP). This includes the maximum number of independent runs (e.g., plates) that can be performed by an analyst in a fixed period of time.
但必须注意,为获得具有代表性的方法中间精密度(IP)估算,验证应按照开发期间设定的限制条件(或作为方法 SOP 的一部分)执行。这包括分析人员在固定时间内可完成的独立运行(如培养板)最大数量。
2.2 Bioassay Validation Protocol 生物检定验证方案
A bioassay validation protocol should include the validation design with between-run factors (and within-run if these interact with between-run factors), the number of validation runs, the design layout (crossed or nested), the validation parameters with justified acceptance criteria, and a proposed data-analysis plan.
生物检定验证方案应包含:纳入运行间因素的验证设计(若运行内因素与运行间因素存在交互作用,也应纳入)、验证运行次数、设计布局(交叉或嵌套)、带有合理性论证的可接受标准的验证参数,以及拟定的数据分析计划。
The validation might be formulated based on TAE rather than on individual parameters (see
3.5 Total Analytical Error
). Use of TAE should be clearly specified in the protocol as the primary basis of evaluating fitness for use or as an adjunct to a parameters-based validation.
验证可基于 ** 总分析误差(TAE)** 而非单个参数进行设计(见 3.5 总分析误差)。在方案中应明确规定将 TAE 作为评估适用性的主要依据,或作为基于参数验证的辅助手段。
A brief summary of the method SOP (the bioassay principle, design, and data analysis) is useful to justify possible sources of variability (e.g., potential ruggedness factors). System and sample suitability criteria should be established beforehand as part of design of the bioassay method. Because these may be based on limited data and a limited collection of samples, they may be proposed as tentative and can be updated based on results from the validation (or after suitable experience during routine testing). However, the suitability criteria should not be changed if, during development, these have been established to limit impact on relative potency determination (i.e., have been established to ensure accuracy or precision). They might be changed if they are based instead on variability of the suitability parameter rather than impact, and the change is supported by other measures of acceptable performance of the method.
对方法 SOP 的简要概述(生物检定原理、设计及数据分析)有助于论证潜在的变异来源(如潜在的稳健性因素)。系统与样品适用性标准应作为生物检定方法设计的一部分预先建立。由于这些标准可能基于有限的数据和样品集合,可拟定为暂定标准,并可根据验证结果(或在日常检验中获得充分经验后)进行更新。但是,若在开发期间已确定这些标准用于限制对相对效价测定的影响(即为确保准确度或精密度而设定),则不得修改。若这些标准仅基于适用性参数的变异而非其影响,且变更得到方法可接受性能的其他指标支持,则可进行修改。
Action should be prespecified when a validation run fails to meet a suitability criterion. As these should be rare, it is acceptable to remove the run without replacement. Note that this might result in an imbalanced design, requiring special software or statistical support to analyze the remaining validation data, and will reduce the statistical power if the validation uses a small number of runs. Note also an abundance of failed validation runs (more than is predicted by the method for deriving suitability criteria) should be regarded as a validation failure.
应预先规定当验证运行不满足适用性标准时的处置措施。由于此类情况应较少发生,可删除该运行且不补充。注意这可能导致设计不均衡,需要专用软件或统计支持分析剩余验证数据,且若验证运行次数较少,会降低统计效力。同时需注意,若验证运行失败数量过多(超出建立适用性标准方法的预期),应视为验证失败。
Inability to meet validation acceptance criteria may result in validation failure, a limit on the range of potency that can be measured in the bioassay, or a modification to the release procedure format (i.e., the replication strategy) to achieve the desired precision in RV.
不满足验证可接受标准可能导致验证失败、限定生物检定可测定的效价范围,或修改放行程序模式(即重复策略)以实现可报告值(RV)的预期精密度。
2.3 Bioassay Validation Sample Size 生物检定验证样本量
The application of statistical tests, including the assessment of conformance of validation parameters to their acceptance criteria, involves risks. One risk is that the validation result does not meet its acceptance criterion although the property associated with that result is satisfactory (called a type II error for an equivalence test; also, producer’s risk or false negative); another, the converse, is that the validation result meets its acceptance criterion although the property is truly unsatisfactory (called a type I error for an equivalence test; also, consumer’s risk or false positive).
统计检验的应用(包括评估验证参数是否符合可接受标准)涉及风险。一种风险是尽管结果对应的属性实际合格,但验证结果不满足可接受标准(等效性检验中的 II类错误,亦称生产者风险或假阴性);另一种相反风险是尽管属性实际不合格,但验证结果满足可接受标准(等效性检验中的 I 类错误,亦称消费者风险或假阳性)。
The two types of risk can be simultaneously controlled via determination of the number of runs (and within-run replicates) to be conducted in the validation. Details for how this is done are reserved for
Appendix A—Bioassay Validation Example
.
可通过确定验证中执行的运行次数(及运行内重复次数)同时控制这两类风险。具体实施方法详见附录 A—— 生物检定验证示例。
2.4 Bioassay Validation Data Analysis 生物检定验证数据分析
A thorough analysis of the validation data should include graphical and statistical summaries, including validation parameter estimates (including CIs) and a conclusion regarding success or failure to meet their acceptance criteria. The analysis should follow the specifics of a data-analysis plan outlined in the validation protocol. Data should be analyzed in accord with the scale (e.g., log transformed) determined during development and specified in the method SOP (see
Appendix C—Data and Statistical Considerations, C1. Scale of Analysis
).
对验证数据的完整分析应包括图表与统计汇总,其中包含验证参数估算值(含置信区间 CI)以及关于是否满足可接受标准的结论。分析应遵循验证方案中概述的数据分析计划细则。数据应按照开发期间确定并在方法 SOP 中规定的尺度(如对数转换)进行分析(见附录 C—— 数据与统计考量,C1 分析尺度)。
Analyses are typically performed on data at each validation level. This includes VC analysis to estimate the contributions of within- and between-run (or individual factor) sources of variability to the bioassay IP and ANOVA to estimate RB and its associated CI. Linearity is assessed across levels using regression analysis.
通常在每个验证水平下对数据进行分析。这包括用于估算运行内与运行间(或各独立因素)变异来源对生物检定中间精密度(IP)贡献的方差成分(VC)分析,以及用于估算相对偏倚(RB)及其置信区间(CI)的方差分析(ANOVA)。采用回归分析评估各水平间的线性。
Statistical success requires evidence of conformance of an estimated parameter to its acceptance criterion. It is not sufficient to fail to find a significant departure from a target (e.g., RB = 0%; sometimes called a difference test). Instead, an equivalence (or noninferiority) test using a CI or CB should be employed to show that a validation parameter is likely to be within appropriately justified acceptance criteria (see
〈1010〉 Appendix 3: Equivalence and Noninferiority Testing
).
统计合格需要有证据证明估算参数符合其可接受标准。仅未发现与目标值存在显著偏离(如 RB = 0%,有时称为差异性检验)并不充分。相反,应采用基于置信区间(CI)或置信边界(CB)的等效性(或非劣效性)检验,以证明验证参数极有可能在经过合理论证的可接受标准范围内(见〈1010〉附录 3:等效性与非劣效性检验)。
An equivalence approach for assessment of relative accuracy will be illustrated in
Appendix A—Bioassay Validation Example
. An upper CB on IP can be used as statistical evidence of noninferiority (relative to the acceptance criterion) while prediction intervals are used with TAE. These may impact validation study sample size and will be illustrated as an "Advanced Consideration" in
Appendix A—Bioassay Validation Example
.
附录 A—— 生物检定验证示例中将举例说明用于评估相对准确度的等效性方法。中间精密度(IP)的单侧置信上限(CB)可用作非劣效性的统计证据(相对于可接受标准),而预测区间与总分析误差(TAE)结合使用。这些可能影响验证研究样本量,并将在附录 A—— 生物检定验证示例中作为 “高级考量” 进行说明。
If specified in the data analysis plan, MEM can be used to analyze validation data across validation sample levels while TAE might be proposed instead of the individual parameter approach.
若数据分析计划中有规定,可采用模型辅助验证法(MEM) 跨验证样品水平分析验证数据,也可提出采用 TAE 替代单个参数方法。
2.5 Documentation of Bioassay Validation Results 生物检定验证结果文件记录
Bioassay validation results should be documented in a bioassay validation report. The report should include the raw data and intermediate results (e.g., VC estimates) that facilitate reproduction of the bioassay validation analysis and the design of procedures using the bioassay method. Estimates of validation parameters should be reported at each validation level and overall if planned. Deviations from the validation protocol should be documented with justification. The conclusions from the study should be clearly stated with references to follow-up action(s) as necessary. Follow-up actions can include amendment of system or sample suitability criteria as well as a change to the release procedure format. Since the change in format is a mathematical function of the intermediate validation results there is no need for revalidation after a format change.
生物检定验证结果应记录在生物检定验证报告中。报告应包含原始数据与中间结果(如方差成分估算值),以便于重现生物检定验证分析并设计采用该生物检定方法的程序。应报告每个验证水平下的验证参数估算值,若有计划还应报告总体估算值。偏离验证方案的情况应附带合理性说明文件化。应清晰陈述研究结论,并在必要时注明后续措施。后续措施可包括修订系统或样品适用性标准以及变更放行程序模式。由于模式变更是基于中间验证结果的数学函数,因此模式变更后无需重新验证。
It is good practice to include results of other studies used to build validity into the bioassay method (e.g., robustness studies) or of other validation parameters (selectivity and specificity) in the final validation report.
良好规范是在最终验证报告中纳入用于支持生物检定方法有效性的其他研究结果(如耐用性研究)或其他验证参数(专属性与特异性)。
2.6 Continued Performance Verification 持续性能确认
Once a bioassay has been validated it can be implemented for routine testing of late development and commercial product materials. However, in keeping with a life cycle approach, it is important to monitor its behavior over time (Stage 3, continued performance verification; see 〈1220〉). This is partly accomplished using SPC charts (see 〈1010〉) on suitability parameters including potency of control samples. The purpose of these charts is to detect an aberrant run that might impact potency determination of samples tested in that run or a trend (i.e., a shift or drift) in method performance over time. If a trend is observed in an SPC chart, the cause should be investigated. If an assignable cause has been identified and resolved, the SPC limits may be reevaluated to address future abnormal behavior in bioassay method performance.
生物检定一经验证,即可用于后期开发与商业化产品的日常检验。然而,遵循生命周期理念,持续监测其随时间的表现至关重要(第 3 阶段:持续性能确认;见〈1220〉)。这可部分通过对适用性参数(包括对照样品效价)建立统计过程控制图(SPC) 实现(见〈1010〉)。控制图的目的是识别可能影响该次运行中样品效价测定的异常运行,或方法性能随时间的趋势(即偏移或漂移)。若在 SPC 图中观察到趋势,应调查原因。若已识别并解决可归属原因,可重新评估 SPC 限度,以应对未来生物检定方法性能的异常表现。
It is important to note that SPC monitoring of bioassay performance should be established early in the bioassay life cycle. Early establishment of processes for monitoring bioassay performance helps assure the link of potencies determined during development to testing of commercial product.
必须注意,生物检定性能的 SPC 监测应在生物检定生命周期早期建立。尽早建立监测流程有助于确保开发期间测定的效价与商业化产品检验结果的关联性。
Routine analytical control can be linked to life cycle events by collecting appropriate metadata such as critical reagent sources or lots, analysts, real time levels of critical conditions, and other factors that may influence bioassay performance. Collection of such data facilitates product or assay investigations and can be used to improve knowledge about factors that impact bioassay performance.
日常分析控制可通过收集适当的元数据(如关键试剂来源或批次、分析人员、关键条件的实时水平及其他可能影响生物检定性能的因素)与生命周期事件关联。收集此类数据有助于产品或检定方法的调查,并可用于深化对影响生物检定性能因素的认知。
Continuous performance verification should also include maintenance associated with planned changes during routine use of a bioassay method (i.e., post-approval changes). This includes bioassay method transfer, standard qualification, and method bridging. Planned changes should be supported by comparability protocols that are designed to assess risks to patients as well as manufacturing and supply.
持续性能确认还应包括与生物检定方法日常使用期间计划变更相关的维护(即批准后变更)。这包括生物检定方法转移、标准品标定及方法桥接。计划变更应得到可比性方案的支持,可比性方案旨在评估对患者以及生产与供应的风险。